Open Access
Research Article
Max Screen
ISSN: 2456-5482
Copyright: © 2019 de Alba Iriarte B. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Canavan disease, a genetic and metabolic neurodegenerative disorder, occurs at early ages, causing visual, neurological alterations, and fatal consequences. There is no curative treatment, although lithium citrate is being investigated. This is the first case reported in the Basque region of Spain and it introduces a novel mutation to Europe.
Keywords:Aspartoacylase; Canavan; Leukodystrophy; N-acetylaspartate; Neurodegenerative
List of abbreviations:CD: Canavan Disease; MRI: Magnetic Resonance Imaging; NAA: N-acetylaspartate
Canavan disease (CD) or aspartoacylase deficiency is a rare genetic and metabolic neurodegenerative disorder (1:100,000 live births) which is transmitted by autosomal recessive inheritance [1]. The disease is caused by mutations in the ASPA gene (17p13.2), which encodes aspartoacylase [2]. The deficiency of that enzyme leads to an accumulation of N-acetylaspartate in the brain tissue, resulting in oligodendrocyte dysfunction, spongiform changes and absence of myelin. It occurs at early ages, causing visual and neurological alterations. Generally, symptoms begin between the 3rd and 6th month of life. Patients present severe hypotonia, macrocephaly, abnormal visual behavior, severe developmental delay and other neurological disorders [3-6]. Accurate diagnosis is essential due to the devastating course. Nowadays there is no effective treatment for this disease and patients usually die during childhood. Radiological tests, metabolic analysis and genetic study must be used together in order to make a certainly diagnosis [7].
CD is one of the most prevalent degenerative cerebral diseases of infancy. It is more common among Ashkenazi Jews (1:6,400- 13,500) but in other populations has been discovered as well [1]. However, there is not enough data to estimate the prevalence [3]. In European continent and in our environment, as well, is unknown. The authors report the first case of Canavan disease in Basque Country associated to a novel mutation which is identified for the first time in Europe.
The patient of our study is a 2-year-old boy from the Gipuzkoa province in the Basque region of Spain. He was full term after an uneventful pregnancy and his parents are healthy but consanguineous, cousins, of Pakistani origin. At 3rd month of age he was hospitalized due to abnormal visual behavior. His head circumference was 42 cm (50th percentile), length was 60 cm (10th-25th percentile) and weight was 5640 g (10th percentile). In the physical examination was observed hypertelorism and plagiocephaly, and presented poor visual contact, abnormal external eye movements, occasional nystagmus, lacked head control, mild axial hypotonia with hypertonia of the lower limbs and bilateral Babinski sign. Our patient was subjected to different complementary tests to obtain the diagnosis.
Cranial ultrasound revealed an increased echogenicity of the gray nuclei of the base and the brainstem, and cranial MRI showed signal alteration with marked restriction of diffusion of cerebellar, brainstem and basal ganglia white matter, which are typical characteristics of leukodystrophy (Figure 1). Because of the absence of hypoxic-ischemic background and the affectation being bilateral and symmetrical, a metabolic disease was considered as the first possibility.
Biochemistry, hemogram, gasometry and serology samples were processed without notable alterations in the results. The profile of plasma, urine and cerebrospinal fluid amino acids showed normal results. Organic acids in urine were performed by gas chromatography-mass spectrometry (GC-MS) yielding two huge peaks of 2 TMS and 3 TMS N-acetylaspartate (NAA) with a medium value of 1527 mmol/mol creatinine [reference value < 36] (Figure 2). The result was compatible with the diagnosis of Canavan disease.
The diagnosis was confirmed by analysis of the mutations in ASPA gene. Genomic DNA was extracted from peripheral blood leukocytes using a MagNA Pure LC automated system (Roche) following manufacturer´s instructions. All exons and flanking intronic regions of the ASPA gene (NM_000049.3) were amplified by standard polymerase chain reaction. PCR products were sequenced using the cycle sequencing BigDye Terminator v3.1 in both forward and reverse directions. Base and variant calling were performed using the SeqScape v2.5 software (Applied Biosystems, Foster City, CA).
The child presented the mutation c.162C>A (p.Asn54Lys) in homozygosis in exon 2 of the ASPA gene. To discard that the apparent homozygosis is due to the presence of a deletion in one of the copies of the ASPA gene, the status of carriers of both parents should be checked. The parents showed the same variant in heterozygosis. It has been verified, by targeted Sanger sequencing method, the status of heterozygous carrier in both parents, demonstrating that the patient is homozygous for the mutation (Figure 3).
Canavan disease is a rare metabolic neurodegenerative autosomal disorder that is recessively inherited and has unknown prevalence in our country and in Europe [3]. There have been few studies in Europe, but none reported the mutation c.162C>A (p.Asn54Lys).
CD is one of the most common degenerative disorders of the brain in childhood, which occurs at an early age, causing visual and neurological alterations [8]. It has serious consequences for the patients. Although these children appear normal at birth, certain developmental milestones and symptoms like muscle tone deficiency and poor head control are noticeable [4], becoming later in a serious neurological disease characterized by leukodystrophy and macrocephaly [9], which causes feeding problems and loss of vision. The delayed development is more evident between the 3rd-6th months of life (not reaching seating, with poor purposeful use of hands and little communicative intention). The head circumference is normal at birth, increasing between 6 months-1 year above the 90th percentile. As they grow up, the hypotonia is replaced by spasticity, with rigidity in the limbs and trunk. They are often irritable and can suffer sleep disorders, seizures and blindness due to optic atrophy. Hearing usually does not deteriorate.
Canavan disease belongs to the group of Central Organic Acidurias of the Innate Errors of Metabolism (IEM) that affects especially to the brain [3]. The accumulation of N-acetylaspartate in brain results in N-acetylaspartic aciduria, causing a severe and progressive cerebral atrophy of the white matter that is appreciated in neuroimaging, being helpfully and specific for Aspartoacylase deficiency [9,10]. Leukodystrophy with alterations of the white matter are characteristic features on cranial MRI. The most severe abnormalities are present in the subcortical white matter being central white matter structures well preserved. In the differential diagnosis have to be into account other neurodegenerative disorders of childhood associated with a normal or large head size or spongy brain degeneration: Alexander disease, Tay-Sachs disease, metachromatic leukodystrophy, glutaric acidemia type 1, Leigh syndrome, Non-Ketotic Hyperglycinemia (NKH) encephalopathy and viral encephalitis [8]. In the past the diagnosis of Canavan disease was established measuring aspartoacylase enzyme in cultured skin fibroblasts, but the demonstration of hundredfold elevations of NAA in urine organic acids analyses performed by GC-MS is now more practicable and fast being pathognomonic [11,12].
Aspartoacylase deficiency is caused by mutations in the ASPA gene (17p13.2), which encodes aspartoacylase [2]. The disease is present worldwide and can occur in any ethnic group, although it is more common among Ashkenazi Jews [1]. The genetic test for the mutation of the ASPA gene must be offered to the families of Ashkenazi Jews. More than 100 pathogenic variants have been described in ASPA gene to date, of which 78 are single nucleotide variants (HGMD® Professional 2019.1 database). Three of them are the most frequent [8]. The mutations p.Glu285Ala and p.Tyr231Ter represent 98% of pathogenic variants in Ashkenazi Jews individuals and 3% in non-Ashkenazi Jews. Another mutation, p.Ala305Glu, represents 30-60% of pathogenic variants in nonAshkenazi Jews individuals and approximately 1% in Ashkenazi Jews. The other mutations discovered are less known.
The child presented a c.162C>A (p.Asn54Lys) variant in homozygosis and his parents are heterozygous carriers of that variant. The nucleotide change present in exon 2 of the ASPA gene presumably causes as asparagine to lysine substitution at codon 54, previously reported as pathogenic. The mutational study confirmed the diagnosis of Canavan disease for the first time in the Basque region of Spain and identified an unknown mutation of this disease only previously reported twice in two other parts of the world. After reviewing the literature, although several cases of the disease have been found in the European continent [13], none reported the mutation described in our case becoming the first case of Canavan disease associated to the mutation c.162C>A (p.Asn54Lys) in Europe.
This mutation has been previously reported for the first time in Australia in 2004, in the Ashkenazi Jews [14] and in 2012, in Punjab (Pakistan) in a case of a 2-year-old child of consanguineous parents of non-Jew descent [12]. The origin of the family of our study is from Pakistan and the case looks like the second case reported.
The Ashkenazi Jews are a population distributed in Central and Eastern Europe, North America and Oceania. The mutation identified in our case is frequent in the Asian continent, but in view of the increasing transnational immigration, sometimes with a greater degree of consanguinity, we must suspect this type of pathology in the presence of the symptoms described and take into account that the mutation c.162C>A (p.Asn54Lys) may be the cause of Canavan disease.
Currently, there is no cure for this disease, and management of these patients consists mainly on treating symptoms. Although death often occurs before 18 months of age, some patients can live to adulthood thanks to the supporting treatment. Nowadays, there is no known or effective treatment for the disease [12], but upcoming studies using gene therapy [15,16], enzyme replacement therapy [17] or other drugs which may offer hope for the future. Amongst the drugs that are being researched, glycerol triacetate has not shown any improvements [18]. Lithium citrate, instead, seems to improve visual contact and social interaction in some examples [19,20]. There have been described cases in which after 1 year of treatment with lithium citrate NAA levels decreased by approximately 20% in the brain region and 80% in urine improving visual follow-up. Side effects during treatment are mild and rare, but can involve multiple systems, particularly the central nervous system (causing tremors, headache and dizziness) and the renal, gastrointestinal and endocrine systems. Most of the adverse effects are dose-dependent, therefore lithium dosage should be closely monitored and renal function, cardiac function and thyroid function should be re-assessed periodically.
Consequently, it is demonstrated that due to the uncommon adverse effects and limited treatment options, lithium citrate can be a good therapeutic alternative to slow the progression of the disease and improve the quality of life of patients, until the gene therapy is effective.
Our patient, after 18 months of treatment with lithium citrate, has been mostly stable over the follow up period with minor gains in motor or language categories. During monitoring, neurologic examination was performed every two months. Over the first months of lithium treatment, the patient showed slight improvement in eye contact, followed by a progressive deterioration of visual contact and persistence of erratic eye movements. No motor improvement has been observed, remaining cervicoaxial hypotonia with spasticity and hypertonia of all extremities. No seizures have been observed. Due to swallowing problems the patient has required the placement of a gastrostomy button and he is under inhaled corticosteroid therapy because of recurrent bronchitis. The decrease of NAA after lithium treatment has not been studied in our patient.
In the studied patient lithium was well tolerated. Lithium serum levels were in the therapeutic range during most of the treatment period, without noticing any adverse effect. There were no abnormalities in renal, thyroid or liver function. In addition to pharmacological treatment, our patient is regularly attending a specialized center for children with cerebral palsy, where he receives physiotherapy and occupational therapy, among others.
It is necessary to diagnose CD as soon as possible, carry out a study of family carriers and offer genetic counseling to the family, including the possibility of prenatal diagnosis in another gestation. Genetic counseling is recommended for future parents who have a family history of Canavan disease, especially if both parents have Ashkenazi Jewish ancestry
The authors report the first case of Canavan disease diagnosed in the Basque region of Spain in a child of consanguineous parents of Pakistani origin. Symptoms and data from radiological tests, metabolic analysis and genetic study let us to establish a certainly diagnosis. In the genetic study we found out a mutation not known previously in Europe. Given the limited treatment options, initiating treatment with lithium citrate should be considered in these patients in order to improve their visual contact and social interaction, with few adverse effects.
Thanks to Dr. José Julián Landa Maya (Pediatric Department), Dr. Adolfo Garrido Chércoles (Clinical Biochemistry Laboratory), Dr. Raquel Sáez Villaverde (Genetics Department), Dr. María Elena Redín Sarasola (Clinical Biochemistry Laboratory) and Donostia University Hospital.

![]()

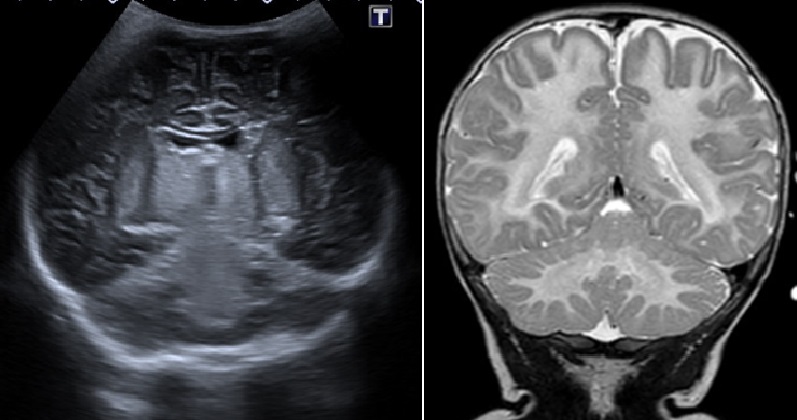 |
| Figure 1: Cranial ultrasound (left) showing an increased echogenicity of the gray nuclei of the base and brainstem. Cranial MRI (right) showing leukodystrophy with marked restriction of diffusion of cerebellar, brainstem and basal ganglia white matter |
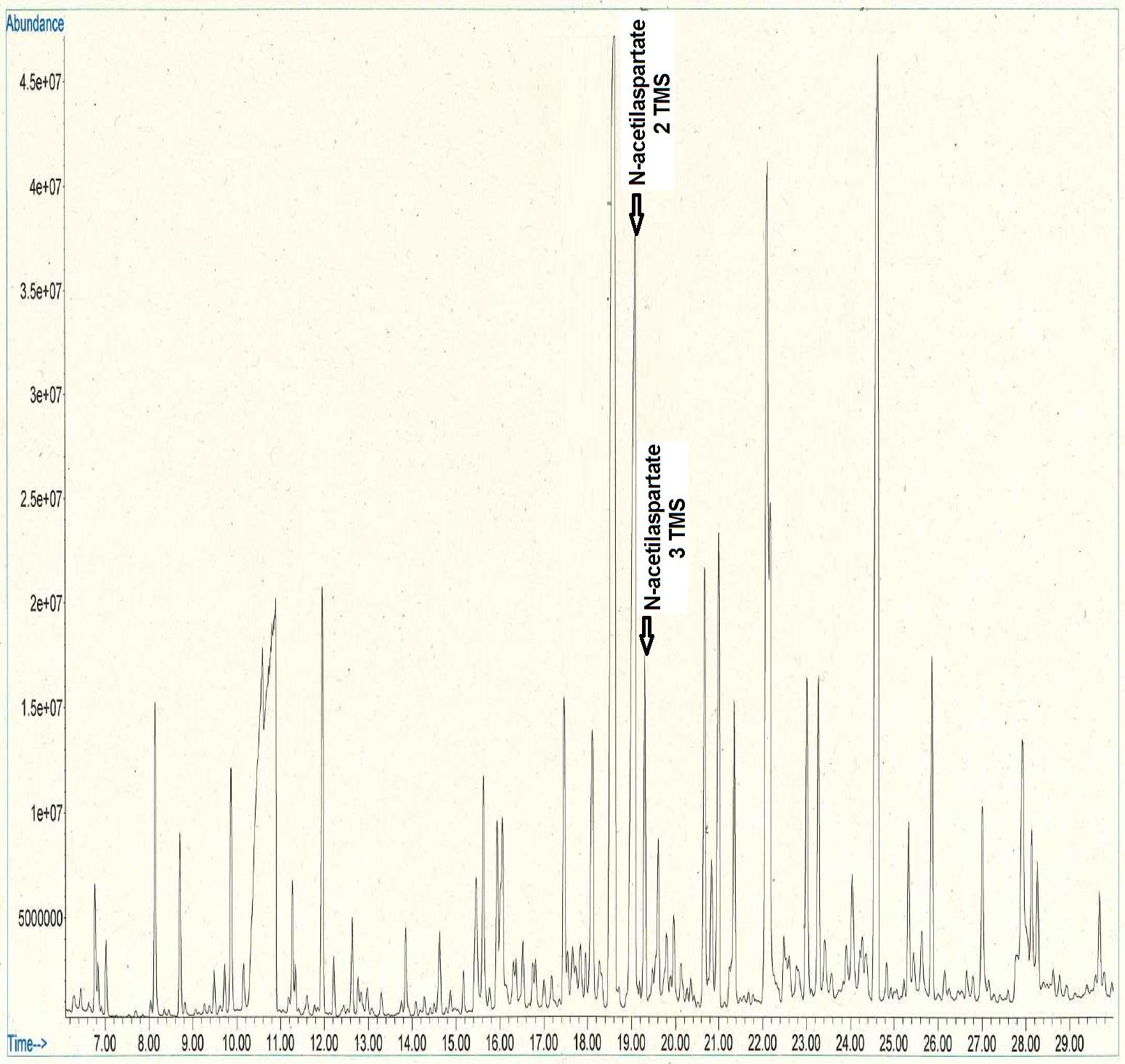 |
| Figure 2: Increased NAA by GC-MS, compatible with diagnosis of Canavan disease |
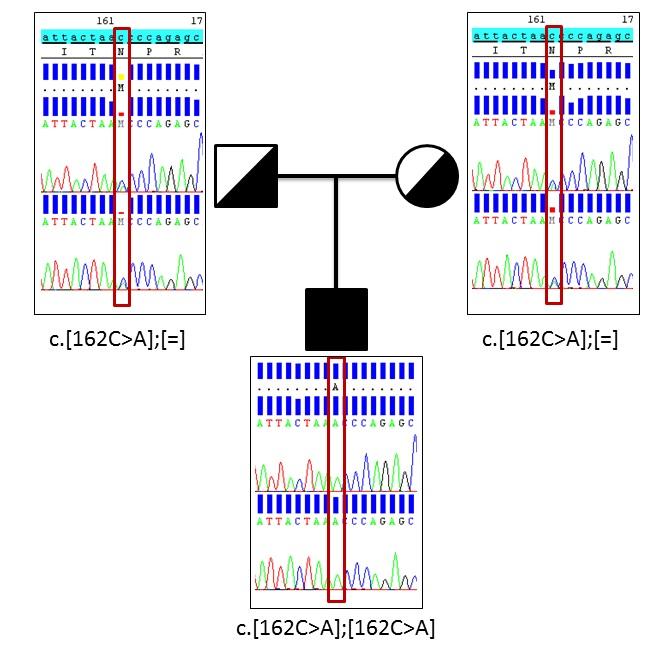 |
| Figure 3: Sanger sequencing of missense variant in ASPA gene |




































