Open Access
Research Article
Max Screen
Copyright: © 2022 Gomes RR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Progressive supranuclear palsy (PSP) is a rare neurodegenerative parkinsonian syndrome with distinct clinical features which tends to be progressive, causing vertical supranuclear gaze palsy, frontal lobe cognitive decline, postural instability and progressive axial rigidity. Clinical examination typically reveals ocular motor dysfunction including restricted vertical gaze, slow vertical saccades and “eyelid opening apraxia” with intact vestibule ocular reflex. PSP falls under the rubric of Parkinsonism plus syndromes that are a group of heterogeneous degenerative neurological disorders that differ from the classical idiopathic Parkinson’s disease. PSP is often underreported, making it important for clinicians to be aware of this disorder. Here we report a case of PSP, which presented primarily with speech disturbances and recurrent falls due to postural instability. On investigating, the magnetic resonance imaging (MRI) revealed a classical sign diagnostic of PSP. This teaching case report describes management and prognosis of the disease.
Keywords: Progressive Supranuclear Palsy, Axial Rigidity, Parkinsonism Plus, Postural Instability
Progressive supranuclear palsy (PSP) is a rare neurodegenerative disease affecting the brain stem, basal ganglia, and cerebellum [1]. First described in 1964 by Steele et al., PSP had been referred to historically as Steele-Richardson-Olszewski syndrome [2]. They reported nine cases with the aforementioned findings, which veered from the typical presentation of idiopathic Parkinson’s disease [2]. Since its initial characterization, PSP has been further categorized into different phenotypes with notable overlap with other neurodegenerative disorders. Early in the onset of the disease, patients with PSP are often misdiagnosed as having idiopathic Parkinson’s disease. Although the exact cause of this disease is not known, evidence suggests it is due to abnormal deposition of tau protein in neuronal tissues, thereby making it a tauopathy. PSP is a disorder of middle or late age, affecting both men and women almost equally after the sixth decade of life, with a prevalence rate of 5-6 per 100,000 and median survival of 7-12 years after diagnosis [3]. The presentation may vary from complaints of multiple falls directed backwards, dysarthria, downward gaze palsy, axial rigidity, and eventually cognitive impairment [1,3]. Magnetic resonance imaging (MRI) is helpful in making the diagnosis. Here, we report a case of early onset PSP presenting with repetition of words, abnormal eye movement, and repeated falls. Given that the average life expectancy following diagnosis is 5-10 years, it is imperative for practitioners to identify and manage the disease appropriately [4,5].
A 60-year-old Muslim, lifelong nonsmoker retired post man from rural Bangladesh, not known to have diabetes, hypertension, bronchial asthma or ischemic heart disease, presented to the outpatient department with repetition of words spoken to him, progressive declination of memory, slurring of speech and frequent fall at home with onset 3 years history prior to presentation. He also had generalized muscular weakness. The patient was unable to take care of himself compounded with slowness in all activities. He became aloof and did not interact with anyone. He had trouble swallowing during feeding and needed assistance. Sleep and appetite were impaired with no history of tremulousness, hallucinations, fecal and urinary incontinence. Family history was insignificant. No treatment had ever been sought.
On general examination vital parameters were normal. Multiple bruise all over her body due to recurrent falls were noticed while systemic examination was normal. On mental status examination, the patient was conscious, with impaired attention, and he kept repeating the words said to him. No symptoms suggestive of Tourette’s syndrome were present. It was difficult to conduct a formal examination. There was axial rigidity. Routine investigations were within normal limits. MRI brain showed all of hummingbird sign” or “penguin sign (figure 1), Mickey Mouse sign (figure 2) and Morning Glory sign (figure 2).
Treatment provided was mainly symptomatic in the form of citicoline (400 mg twice daily), piracetam (800 mg twice daily), baclofen (30 mg twice daily) for rigidity, and clonazepam (0.5 mg at night) for sleep. Levodopa + carbidopa (110 + 10 mg in two divided doses) was added by the neurophysician. Physiotherapy was started. There was minimal improvement after 4 weeks of treatment. Sleep improved slightly but communication remained poor. The prognosis was explained to the relatives.
PSP is the most common degenerative, atypical parkinsonian disorder, with a prevalence of 6.4 per 100,000 according to Schrag et al. [6,7]. The incidence is reported to increase with age from 1.7 cases per 100,000 at 50-59 years to 14.7 per 100,000 per year at 80- 99 years [8,9]. The mean age of diagnosis is approximately 65 years, with no racial or sex predilection. No significant risk factors for developing PSP have been identified [4]. The clinical research criteria given by the National Institute of Neurological Disorders and Stroke (NINDS-SPS) for the diagnosis of PSP was used to make a diagnosis in this case [2,10].
Although no definitive genetic factors have been identified, recent studies suggest there may be a genetic susceptibility in patients with mutations of the tau gene. PSP is currently considered a “tauopathy” [11]. PSP results from an aggregation of abnormally phosphorylated tau proteins. Tau proteins aid in axonal transport and support neuronal microtubules. Localized accumulation of the irregular tau proteins form what are known as neurofibrillary tangles [12]. In addition to the tauopathy, PSP degenerates dopaminergic neurons and cholinergic neurons, lending to loss of basal ganglia, cerebral cortex and, most clinically characteristic, brainstem structures. Structures within the brainstem that atrophy are the dorsal midbrain, notably the midbrain tegmentum and pedunculopontine nucleus, which lends to postural stability, and the motor nuclei of the cranial nerves in advanced stages of the disease. Given the atrophy of the midbrain tegmentum, the RIMLF is greatly affected, which decreases the presence of vertical EBNs, and an inability to initiate vertical saccades ensues [13].
The diagnostic criteria for PSP, which were revised in 2017 by the International Parkinson and Movement Disorder Society, must include all “basic features”: sporadic occurrence, age 40 years or older at onset of first PSP-related symptom, and gradual progression of PSP-related symptoms. PSP-related symptoms include ocular motor dysfunction, postural instability, akinesia and cognitive dysfunction. The category of ocular motor dysfunction is further stratified to include vertical supranuclear gaze palsy, slow vertical saccades and frequent macro square wave jerks or “eyelid opening apraxia”. In this delineated criterion, the highest level of certainty for each category is defined as vertical supranuclear gaze palsy, repeated unprovoked falls within three years, progressive freezing of gait within three years, and speech or language disorder that presents as some variant of primary progressive aphasia, respectively. Supportive features that may increase diagnostic confidence, but do not alone suggest a diagnosis, include levodopa resistance, dysphagia, photophobia, and hypokinetic, spastic dysarthria [10,14] Clinical forms of PSP have arisen since its original description in 1964. The original cases presented by Steele-RichardsonOlszewski have been classified as Richardson Syndrome (PSP-RS), which is characterized by postural instability, vertical gaze palsy and cognitive dysfunction. Eight other clinical variants have been described based on the severity and nature of their neurological signs [14]. The gold standard for diagnosing PSP is a comprehensive, post-mortem neuropathological examination to identify the presence of neurofibrillary tangles. Because a definitive diagnosis requires a post-mortem exam, it is crucial for an eyecare provider to be able to identify oculomotor restrictions and make appropriate clinical recommendations for the patient’s systemic care.10Patients with PSP typically present with horizontal or vertical diplopia or asthenopia while reading secondary to convergence insufficiency, horizontal or vertical gaze palsy. Patients may also exhibit impaired slow phase responses of their vertical optokinetic response, as well as slowed volitional saccades [7]. In a few studies, abnormal acoustic-startling reflex (orbicularis oculi response to high-intensity stimulation of the median nerve) and apraxia of eyelid opening have been observed in patients diagnosed with PSP [15]. A decrease in blink frequency may cause symptoms of dry eye, including blurred vision, foreign body sensation, burning or irritation. Appropriate evaluation of extraocular muscle motility and near point of convergence as well as slit-lamp examination to evaluate the tear film and corneal integrity are of particular importance [16-19].
From an ocular standpoint, it is important to determine the etiology of a patient’s presenting symptoms of double vision when reading or limitation of gaze rather than assuming such presentations are idiopathic or presbyopia-associated convergence insufficiency.
In assessing these complaints, the clinician must elicit a thorough case history including recent falls or postural changes and memory changes. Additionally, it is critical to carefully assess extraocular muscle movement, saccades, phorias and vergences at distance and near. Forced duction evaluation is particularly valuable in differentiating a mechanical vs. a neurodegenerative or vascular cause. The posterior thalamo subthalamic paramedian artery, which stems from the posterior cerebral artery, supplies the RIMLF. Thus, an infarction of this artery may result in a superior gaze palsy [20]. Neoplasms, particularly pineal gland tumors, may also lead to vertical gaze palsies [20]. Other vertical gaze palsy conditions include Neimann-Pick type C, an autosomal-recessive condition in which cholesterol and lipids accumulate, dorsal midbrain syndrome, Whipple disease, and midbrain infarction [20].
As previously noted, PSP is often misdiagnosed early in its course as Parkinson’s disease. It shows similar symptoms to Parkinson’s, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). Parkinson’s disease is a neurodegenerative disorder with characteristic skeletal muscle tremor, rigidity and akinesia, all of which are features observed in PSP. However, PSP typically involves greater cognitive dysfunction and speech disturbance than Parkinson’s disease. A study by Song et al. assessed patients with MSA, Parkinson’s disease and PSP. Results showed that 73% of patients with PSP had gaze abnormalities, a characteristic that was absent in Parkinson’s disease and MSA [21]. Song also found that patients with PSP and MSA had a poorer response to levodopa, which is a staple treatment for Parkinson’s disease [22].
MSA is defined as an “adult-onset, sporadic, progressive neurodegenerative disease” with “parkinsonian features, cerebellar ataxia, autonomic failure, urogenital dysfunction, and corticospinal disorders” by the Second Consensus Statement on the Diagnosis of MSA. Although difficult to definitively differentiate from PSP, any sign of autonomic dysfunction would steer the diagnosis towards MSA [7,23].
DLB is defined by the 2017 Dementia with Lewy Bodies Consortium as a disease of “progressive cognitive decline of sufficient magnitude to interfere with normal social or occupational functions, or with usual daily activities”. In DLB, patients have “recurrent visual hallucinations that are typically well-formed and detailed,” rapid eye movement (REM) sleep behavior disorder, and “features of parkinsonianism” [24]. Gait abnormalities, gaze apraxia and saccadic dysfunction may be present in both PSP and DLB, but visual hallucinations are a hallmark feature of DLB [25,26].
Various presenting signs and symptoms overlap between PSP and other neurodegenerative disorders, which makes PSP diagnosis reliant on the clinical findings centered around the 2017 Movement Disorder Society criteria. Neuroimaging such as MRI may be a helpful supplement for diagnosis. Various studies report abnormalities on MRI demonstrating midbrain atrophy, third ventricle dilation, T2-periaqueductal hyperintensities and frontal and temporal atrophy. One hallmark feature of PSP on MRI is the “hummingbird sign” or “penguin sign” demonstrating midbrain tegmental atrophy without pontine atrophy, associated with widening of interpeduncular cistern giving the impression of head and body of a humming bird. Other classical MRI signs in PSP are selective atrophy of the midbrain tegmentum with relative preservation of tectum and cerebral peduncles resembling the head of a Mickey Mouse (Mickey Mouse sign) and increased lateral concavity of midbrain tegmentum. (Morning glory sign). Most useful for differentiating PSP from Parkinson’s disease or other atypical Parkinson’s diseases are the midbrain-to-pontine area ratio, which tends to be lower in PSP patients, and the magnetic resonance parkinsonism index (MRPI), which is greater in PSP patients. The MRPI is a value calculated by determining atrophy of the midbrain, superior cerebellar peduncle, pons and middle cerebellar peduncle [27,28]. Another promising diagnostic method is utilizing positron emission tomography scanning to track the tau protein THK5351 and determine the presence of tau aggregates specific to PSP [29]. This diagnostic tool, however, is predominantly used in research and often not clinically performed due to cost constraints. To our dismay, no effective treatment guidelines are available for PSP. A few recent studies have pointed toward the use of rivastigmine for cognitive enhancement and zolpidem to improve sleep, but these studies are anecdotal in nature [30-32]. Patients with PSP often have limited benefit from levodopa therapy, as opposed to patients with Parkinson’s disease. In a study by Williams et al. investigating 91 pathologically confirmed cases of PSP, 32% of patients presented with a response to levodopa, defined as a 30% or greater improvement in symptoms [33]. There are no successful pharmacological treatment options targeting the disease process, and management centers around symptomatic care. A few case reports have demonstrated some efficacy of botulinum toxin injected into the orbicularis oculi for apraxia of eyelid opening, into upper limbs to improve rigidity and into cricopharyngeal muscle for dysphagia [34-37]. Additional intervention from an interdisciplinary care team consisting of speech, physical and occupational therapists is necessary to facilitate greater independent activities of daily living. Following diagnosis of PSP, the optometrist may play a role in the patient’s care, particularly to address diplopia and dry eye symptoms. A study by Reddy et al. found that corneal sensitivity was reduced in patients with PSP, with 71% reporting they did not have dry eyes. However, evaluation of the tear firm revealed reduced tear break-up time compared to the control group [38]. Even without patent report of dry eye symptoms, it is important for the optometrist to manage the patient’s dry eyes with artificial tears and lubricating ointment or gel formulations for severe exposure keratopathy. Although few reports demonstrate alleviation of double vision when reading with “mirror prism,” we found that Fresnel prisms were particularly useful for alleviating the progressive double vision. experienced by the inpatient [39,40]. The role of a multispecialty treatment team is a must in the management of this complex disorder.
PSP is a rapidly neurodegenerative condition with a poor prognosis. A study by Cosseddu et al. evaluated 100 patients with PSP and found that the average disease duration following diagnosis was 8.25 years [4]. Cosseddu found that patients with dementia at the time of diagnosis had a shorter survival time than those without dementia, with no other significant predictors.4 The most common causes of death for patients with PSP are respiratory-related, with the most frequent complication being aspiration pneumonia [41]. Given the high frequency of respiration complications, surgery requiring general anesthesia should be given significant consideration due to the potential for respiratory failure.
PSP often goes underreported or misdiagnosed and better understanding of PSP can help the clinicians identify the condition. The role of neuroimaging as a tool in diagnosis is crucial along with one’s clinical judgment. Although no pharmacologic therapy halts progression of the disease, a multidisciplinary team can provide patients and their caretakers with the tools to maximize quality of life and minimize debilitating symptoms.
None declared

![]()

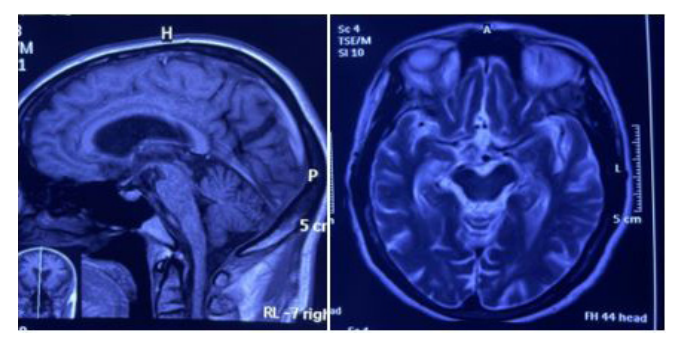 |
| Figure 1 and Figure 2: Mid sagittal T1-weighted MRI of the brain showing midbrain tegmental atrophy without pontine atrophy, associated with widening of interpeduncular cistern giving the impression of head and body of a humming bird(fig 1) and Axial T1-weighted MRI of the brain showing selective atrophy of the midbrain tegmentum with relative preservation of tectum and cerebral peduncles resembling the head of Mickey Mouse and increased lateral concavity of the midbrain tegmentum resembling morning glory(figure 2). |




































