Open Access
Research Article
Max Screen
ISSN: 2348-9812
Copyright: © 2019 Vettumperumal R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
AbstractGround state properties of ATiO3 (A = Ca, Sr, Ba & Pb) pervoskite structures in cubic and tetragonal phase were studied by tight binding linear muffin-tin orbital (TB-LMTO) method in the framework of density functional theory (DFT) with the atomic-sphere approximation (ASA). The total energy of all the compounds come under the above said structures have shown that the cubic phase is the stable structure in the ambient condition. Among these pervoskites maximum bulk modulus was obtained for BaTiO3. Direct (cubic) and indirect (tetragonal) band gap was observed from the band structure calculations and the values fall within the range of 1.5 – 1.7 eV. Electron distribution of each element in the valence and conduction bands was clearly obtained from the density of states (DOS) and partial density of states (PDOS) for all the compounds. The magnetization values were found in the range of 0.4-0.56×10-5μB. The ‘d ’ orbital position of Ti was observed for all the ABO3 compounds and shifted away from the Fermi level except for Ti in BaTiO3. The refractive indices of the pervoskites were calculated from the energy band gap and the value is above 3 for all the compounds.
Keywords: ATiO3 pervoskites (A = Ca, Sr, Ba & Pb); Band structure and density of states; Refractive index and ferromagnetism; Tight Binding Linear Muffin-Tin Orbital Method
Ferroelectrics have wider applications in pyroelectronic detectors, imaging devices, optical memories, modulators, and deflectors. Perovskite materials are an important group of ferroelectrics [1]. The perfect perovskite structure is cubic with a general formula ABO3, where A is a divalent or monovalent metal and B is a tetra- or pentavalent atom. Basically, there are three main features in ABO3 perovskites [2]. In the first kind, the corner linked “O” atoms in the octahedral arrangements with a minor distortion are treated as rigid units to a first approximation. Secondly, inside the octahedron appears the off-centered B-cation which is associated with the phenomenon of ferro or anti-ferroelectricity. The sense of displacement in one-octahedron by way of one corner displacement of the B-cations give rise to a chain of diploes; two corner displacements produce a sheet of dipoles and three corner displacements lead to a single three dimensional dipole [3]. Thirdly, the octahedron may tilt in a variety of configurations and have a considerable effect on the lattice parameters. Titanates, ATiO3,where A=Ca,Sr, Ba, and Pb, exhibit different ferroelectric behaviour’s. It is known that CaTiO3and SrTiO3 are incipient ferroelectrics, whereas BaTiO3 and PbTiO3 are well known ferroelectrics with three phases (tetragonal, orthorhombic, and rhombohedral) for BaTiO3 and one tetragonal phase for PbTiO3 [4].
Investigations have been carried out on BaTiO3to study the structural, dielectric, elastic, and thermal properties [5]. Moreover the infrared and the Raman as well as electron-spin resonance spectra of BaTiO3 were also studied in detail [6-9]. Studies on nanocrystalline powders of BaTiO3 have shown that the tetragonal ferroelectric structure disappears below a critical size of the particle, leading to the cubic phase [10-12]. Investigations on SrTiO3 have been carried out to study the structural, dielectric, optical and elastic properties [13-17]. Infrared and electron paramagnetic resonance spectra of SrTiO3 were also studied in detail [18-20]. Various properties of SrTiO3 have also been investigated in earlier studies [21-26]. Semiempirical MO calculations on a series of ATiO3 (A = Ca, Sr, and Ba) perovskites showed that the bare force constant for BaTiO3 is smaller than that of CaTiO3and SrTiO3 [27]. Local distortions exist even in cubic BaTiO3 and PbTiO3. On the other hand, SrTiO3, which has a structure very similar to that of BaTiO3, has no local distortion [28]. However a systematic theoretical study of the optical properties based upon first-principles band structure calculations is still lacking for the above said compounds which are all come under the roof of pervoskite structures. Also, there is less work focus on the magnetic properties ABO3 pervoskites. In this paper, we have chosen four most common ferroelectric materials and calculate their magnetic properties using tight- binding linear muffin tin orbital (TB-LMTO) method within the Atomic Sphere Approximation. They are compared among each other and the refractive indices are calculated in the light of confirming their application in anti-reflecting coating application.
A series of ATiO3 (A = Ca, Sr, Ba and Pb) perovskite ferroelectric structures are chosen for the present study and the available structural parameters are taken from the literature [29]. We present here the electronic structures and calculations of their magnetic properties using TB-LMTO method. The accuracy of the total energies obtained within the density functional theory, using local spin density approximation (LSDA), in many cases is sufficient to predict which structure at a given pressure has the lowest free energy. Empty sphere approach is introduced in all the cases in order to keep the overlap of atomic spheres within 16%. Band dispersions and density of states (DOSs) are obtained in each case. The calculations are done within the ASA. The basis sets used here comprised of augmented Linear Muffin-Tin Orbitals. Within the atomic spheres, the basic functions, the charge density and the potential are expanded in symmetry-adapted, spherical harmonics together with a radial function. Basis functions up to lmax= 3 for Ba and lmax = 2 for Ti and O were used. The calculations are carried out within the LDA and the radial part of the basis is obtained by solving a Schrodinger-like Kohn-Sham equation in which the scalar relativistic corrections have been incorporated. The von Barth-Hedin exchange potential is used. Brillouin-zone integration has been utilized using the tetrahedron method with a mesh of about 500 symmetry-reduced points. Barium 6p orbital, the oxygen 3s and 3d orbitals are down folded and do not contribute to the dimension of the Hamiltonian (H) and the overlap (O) matrices, but carry a charge. This down folding prevents the appearance of ghost bands.
In the structural optimization, the total energies as a function of reduced volume is found from theoretical calculations for the four ATiO3 (A = Ca, Sr, Ba and Pb) perovskites of cubic and tetragonal phase. The total energy is fitted into the Murnaghan’s equation of state [30].
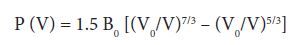
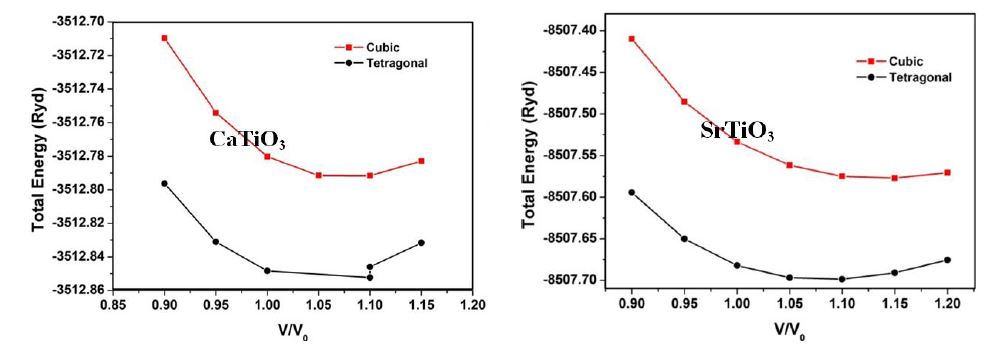
Where, P=-dE/dV and E is the total energy obtained from the band structure calculations and V0 is the theoretically calculated equilibrium volume at ambient conditions. Total energy versus reduced volume is shown in Figure 1. From Figure 1 one can infer that the cubic phase of ATiO3 is stable at ambient conditions and also our observation from calculation predicts semiconducting nature for all the perovskite structures. After fitting the Murnaghan’s equations of state, the bulk modulus of the ATiO3 pervoskites are calculated. The variation of bulk modulus with ionic radius is shown in Figure 2. From the figure, the values of bulk modulus are randomly varied with respect to the ionic radius of ‘A’ cations and maximum value is observed for BaTiO3 in both phases, which indicates the stability of the material. The ground state properties are obtained and presented in Table 1.
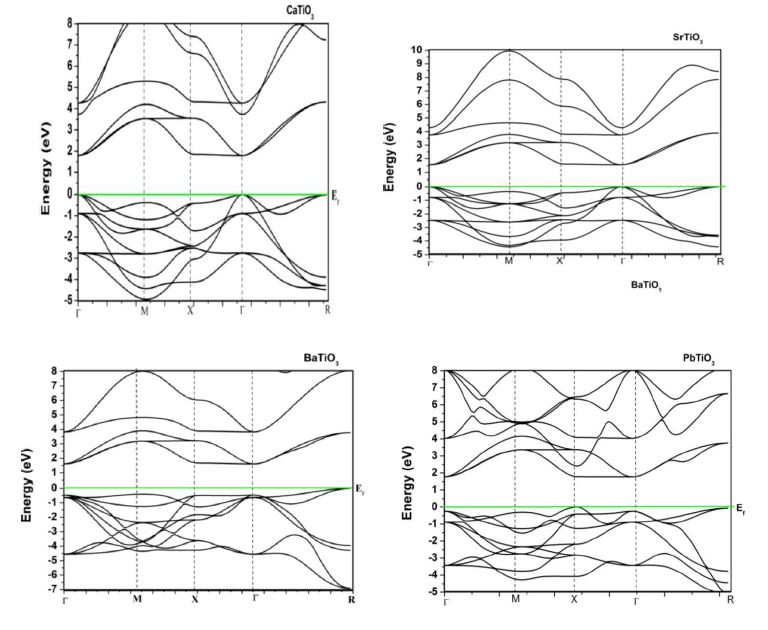
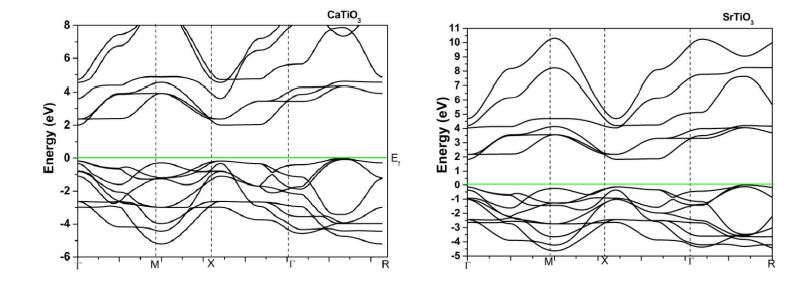
The band structure of ATiO3 (A = Ca, Sr, Ba and Pb) perovskites in cubic and tetragonal phase is obtained by means of LMTO – ASA method. The calculated band structure of cubic (paraelectric) and tetragonal (ferroelectric) phases in the high symmetry directions in the Brillouin zone are depicted in Figure 3. The energy scale is in eV and the origin of energy is arbitrarily set to be at the valence band maximum. The band gap is mainly formed by the interaction between the valence band orbital of O in ‘p’ like states and the antibonding conduction band orbital of Ti and A (Ca, Sr, Ba & Pb) in ‘s’ like states. From the figure, we find a large dispersion of the bands and nine valence bands are derived from ‘2p’ orbitals of oxygen. In cubic phase, these are separated by a direct band gap of 1.5 eV to 1.7 eV whereas in tetragonal phase it predicts an indirect band gap with the same values from the transitional d – derived conduction band. This result is better than the previous TB LMTO-ASA calculations [31]. However, this gap is somewhat lower than the experimental band gap of 3.2 eV [32]. The possibility may be due to the ‘d ’ like orbital of Ti which is deemed to be important for band gap variation owing to p-d interaction. It is believed that the p-d interaction can push the ‘p’ orbital of oxygen upward, thereby inducing the band gap of ATiO3 to become smaller [33]. Also the origin of the band gap discrepancy may be owing to the local density approximation which underestimates the band gaps even for insulators. The valence bands of BaTiO3 in tetragonal phase are slightly changed compared to its cubic phase while other perovskites are not showing any variation in both phases.
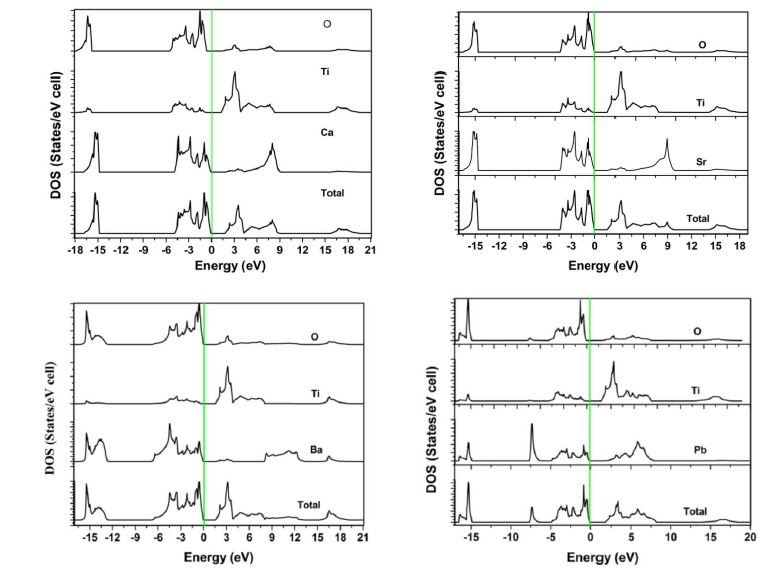
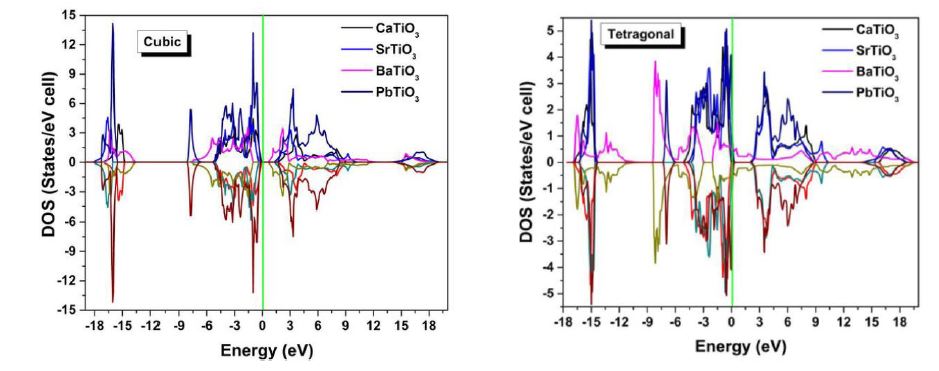
Electron distribution in an energy spectrum is described by the density of states (DOS) and can be measured using photoemission experiment [34]. The total and partial DOS spectrum of ATiO3 (A = Ca, Sr, Ba & Pb) perovskites in both phases are shown in (Figure 4a and b). In these figures, the zero of the energy scale (the top of the valence band) is taken as the position of the Fermi level; the valence and conduction band edges near the Fermi level are quite sharp. All the structures have the same DOS spectrum in the cubic (paraelectric) phase but in the tetragonal (ferroelectric) phase, one more oxygen atom in the valence band side of the DOS spectrum makes an additional contribution. A detailed study of the partial density of states (PDOS) gives information about the contribution of different atomic states in the band structure and also their possible hybridizations. Contribution of the core level states in the valence band is by the presence of O ‘2p’ and A (A = Ca, Sr, Ba & Pb) cations ‘p’ orbitals in both phases. A weak hybridization may occur in the valence band between
‘2p’ orbital of oxygen and ‘p’ orbital of Ca, Sr, Ba & Pb and it lies in the energy range 5.2 eV on top of the valence band. It shows an ionic bond between oxygen and A cations. The presence of ‘d ’ orbital states in the conduction bands of all ATiO3 compounds is felt except for Ba. The ‘d ’ orbital position of Ti in BaTiO3 is much close to the Fermi level compared to all the other perovskite structures. The height of the O ‘2p’ DOS peak is much higher than that of the Ti-3d state peak in both phases. Existence of p-d hybridization is evident from this figure and it also reflects on the Ti ‘3d ’- O ‘2p’ co valency. The observed Ti-d orbital positions are shown in Figure 5. From the figure, Ti-d orbital position is observed to shift away from the Fermi level except for Ti in BaTiO3 and the reason may be due to the contribution of d orbitals of ‘A’ cations in the conduction band. No shift is observed in the tetragonal phase. In order to calculate the magnetic properties, electron spin of the respective elements in each one of the perovskite structure is aligned. The obtained value of magnetic moments is found to be less. The observed values of magnetic moment are in good agreement with the experimental results and presented in Table 1. The spin alignments are shown in Figure 6. The presence of p-d hybridization between O and Ti gives rise to magnetization and the obtained values are confirmed with the PDOS [35].
Refractive index and energy gap of semiconductors represent two fundamental physical aspects that characterize their optical and electronic properties. The device applications of semiconductors as electronic, optical and optoelectronic are very much determined by the nature and magnitude of these two elementary material properties. Empirical relationships modeled by a theoretical numerical analysis have been used to calculate the refractive indices of semiconductors, using their energy gaps. This physical relationship remains strictly intrinsic and specific for the material considered. The model developed stays optimized for the ternary semiconductors because they constitute an intermediate family of semiconductors. As per quantum-mechanics, the model to be developed must obey two constraints. (i) n(Eg) must have a horizontal asymptote equal to 1. (ii) n(Eg) must be hyperbolic. This means that if the band gap Eg increases, the refractive index n decreases, and vice versa, but not linearly. The equation that has been developed by Anani, et al. to calculate the refractive index is,
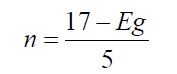
2) Where Eg is the band gap and ‘n’ is the refractive index of the material. The calculated values of refractive indices are listed in Table 1. Exact theoretical derivations of reflection-reducing optical coatings from Maxwell equations and boundary conditions between layer and substrate have been given by Mooney. For anti-reflecting (AR) coating in solar cell applications, the refractive index of the coating should be greater than that of the substrate and in fact, the difference should be as large as possible. Here all the ATiO3 perovskite compounds have higher values of refractive indices. In case of using a glass substrate, the refractive index is 1.5 whereas for ATiO3 the value is above 3. From this result we infer that the ATiO3 (A = Ca, Sr, Ba & Pb) compounds are the best AR materials for glass and for materials of refractive index below 2.
Band structure, magnetic and refractive index of the ATiO3 pervoskite structures were calculated and compared with cubic and tetragonal phases of each compound by TB- LMTO-ASA method. The total energy and bulk modulus of the pervoskite compounds were calculated from the Muranaghan’s equation of state and the stable phase is found to be cubic. All the compounds in cubic phase showed direct band gap whereas tetragonal phase has an indirect band gap. Valence band degeneracy was observed and it shows a semiconductor nature. A weak ionic bond exist between ‘2p’ orbital of oxygen and ‘p’ orbitals of ‘A’ cations and p-d hybridization among the O ‘2p’ and Ti ‘d ’ orbital revealed the covalent bond behaviour as observed from the DOS and PDOS. This p-d hybridization has prompted us to conclude the existence of ferroelectric and ferromagnetic property in the present materials. Observed value of magnetic moments is found to be less. Both phases of ATiO3 structures have higher refractive index values than glass. The results obtained suggest that they can be used for anti-reflecting coating in solar cell applications.

![]()

 |
 |
| Figure 1: Total energy versus reduced volume of ATiO3 pervoskite |
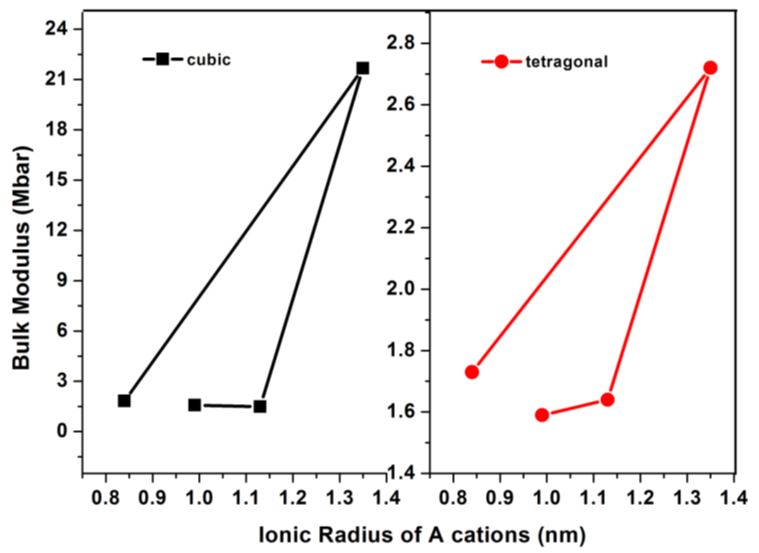 |
| Figure 2: Variation of bulk modulus with respect to A cations ionic radius |
 |
 |
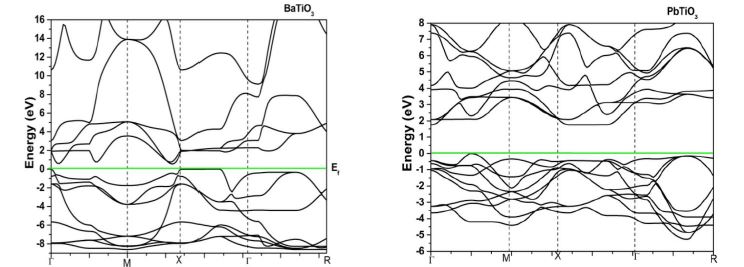 |
| Figure 3: PBand structure diagram of ATiO3 pervoskite (a) cubic (b) tetragonal |
 |
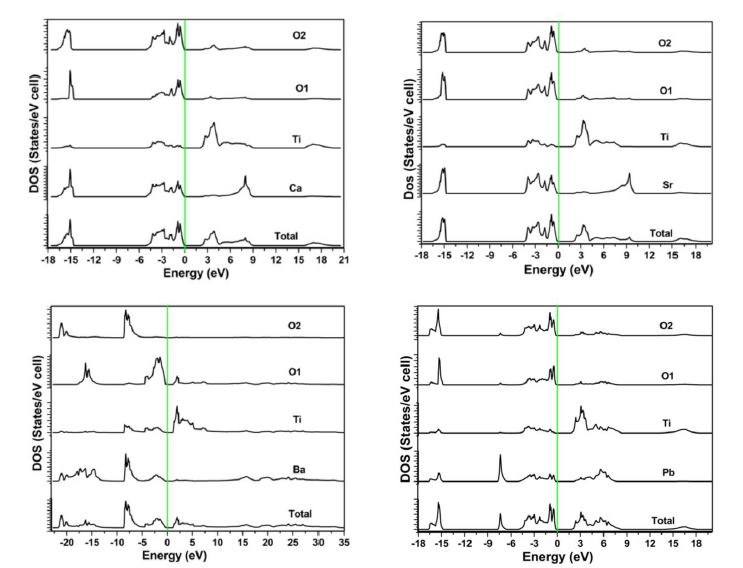 |
| Figure 4: Density of states and partial density of states of ATiO3 pervoskite (a) cubic (b) tetragonal |
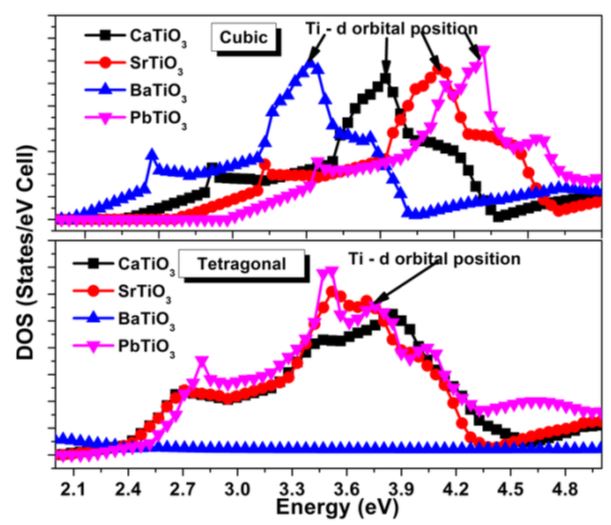 |
| Figure 5: Ti ‘d’ orbital position in cubic and tetragonal phases |
 |
| Figure 6: Electron spins alignment of each element in ATiO3 pervoskite |
Parameters |
Cubic |
Tetragonal |
||||||
CaTiO3 |
SrTiO3 |
BaTiO3 |
PbTiO3 |
CaTiO3 |
SrTiO3 |
BaTiO3 |
PbTiO3 |
|
a (Å) |
7.49 |
7.69 |
7.50 |
7.60 |
3.83 |
3.88 |
3.99 |
3.90 |
c (Å) |
- |
- |
- |
- |
4.14 |
4.05 |
4.036 |
4.15 |
Volume |
427.66 |
456.08 |
423.46 |
439.35 |
428.66 |
448.23 |
450.16 |
449.69 |
Bulk Modulus (Mbar) |
1.57 |
1.49 |
21.67 |
1.82 |
1.59 |
1.64 |
2.72 |
1.73 |
Band Gap (eV) |
1.78 |
1.56 |
1.59 |
1.75 |
1.99 |
1.83 |
1.11 |
1.75 |
Magnetization (x10-5) |
0.38 |
0.45 |
0.52 |
0.56 |
0.42 |
0.41 |
0.56 |
0.52 |
Refractive Index (n) |
3.044 |
3.088 |
3.082 |
3.05 |
3.002 |
3.034 |
3.178 |
3.05 |




































