Open Access
Research Article
Max Screen
ISSN: 2455-7641
Copyright: © 2021 Chukkalore D. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Hb SC disease is a type of hemoglobinopathy that can be viewed as a hybrid of hemoglobin S and C. In this disorder there is co-inheritance of one Hbs gene and one HbC gene resulting in a milder phenotype than sickle cell anemia (SCA). Where as SCA clinical features have been extensively studied, very few studies have been dedicated specifically to HbSC disease as most cases are reported to be silent or of mild severity. As the pandemic continues to evolve with the novel Sars-CoV-2 virus we have learned it can lead to thrombotic complications which could be fatal if not detected early. Of those requiring admission to the intensive care unit, most carry multiple comorbidities (hypertension, diabetes mellitus etc.) leading to worse clinical outcomes. Here, we present a case of a young adult patient with silent hemoglobin SC disease who tested positive for SARS-CoV-2 leading to multiple infarcts, splenic sequestration and respiratory failure. The concurrence of a hemoglobinopathy and COVID-19 should warrant heightened clinical suspicion for unusual outcomes. Hence, providers must remain vigilant while treating any patient with any hemoglobinopathy in the setting of COVID-19.
Keywords: COVID-19; HbSC disease; Acute Splenic Sequestration
Sickling hemoglobinopathies are inherited autosomal recessive disorders that result in production of an abnormal form of hemoglobin. Hemoglobin SC disease is an inherited variant in which individuals have two abnormal beta chains, βS and βc with no hemoglobin A. Hemoglobin SC disease makes up almost 30% of cases of sickle cell disease in the United Kingdom and the United States1. In some regions of West Africa, where the HbC variant arose, HbSC may form more than half of all cases of sickle cell disease [1]. Globally more than 50,000 babies with HbSC are born each year [1]. Although 30% of patients with SCD in the US and UK have HbSC, there is not enough disease-specific data available for solely HbSC thus most data is based on pathogenesis of SCA. HbSC red blood cells (RBCs) contain roughly equal amounts of HbC and HbS and low (1–3%) levels of HbF [2]. The pathogenesis is modulated by interactions between abnormal HbS and HbC which result in red blood cell changes from their usual biconcave disc shape to a sickle shape during de-oxygenation. Upon re-oxygenation, the red blood cell initially resumes its normal shape but, after repeated cycles of “sickling and unsickling,” the red blood cell is permanently damaged and hemolyzes. With the presence of abnormal hemoglobin C which mostly drives this disease, red blood cells are unstable and broken down more quickly than normal and results in enhanced potassium and chloride co transport which causes loss of intracellular potassium resulting in polymerization and further sickling of red blood cells [3]. These sickled cells restrict vascular blood flow and can result in the clinical manifestations of the disease which worsen on dehydration. Curiously, whereas SCA clinical features have been extensively studied, very few studies have been dedicated specifically to HbSC disease. Of those few reported, clinical complications were painful vaso-occlusive crisis, silent infarcts, acute chest syndrome, peripheral neuropathy, priapism, stroke, pulmonary hypertension, leg ulcers, osteonecrosis, infection and an increased emphasis of prevalence on viscosity-associated otological and ophthalmological disorders compared to SCA [4]. HbSC disease is considered to be a variant of SCA, sharing similar clinical complications with a somewhat milder severity and a lower frequency with a life expectancy of 20 years longer [5]. Nevertheless, stroke, retinnitis proliferans, osteonecrosis, and acute chest syndrome have equal or higher incidence in HbSC disease compared to SCA which is likely due to HbC enhancing the formation of intracellular polymer of HbS by dehydrating red cells resulting in thrombotic complications [3]. Here we present a case of young woman with hemoglobin SC disease with severe vaso-occlusive crises precipitated by COVID-19 infection leading to life threatening complications.
A 29-year-old Caucasian female patient with reported history of “sickle cell trait” manifesting as mild pain related crises with no history of prior hospitalizations was evaluated by her primary care physician for subjective fevers, chills, intermittent left lower quadrant abdominal pain and was incidentally diagnosed with COVID-19 via nasopharyngeal PCR. However, at that time she was well appearing, afebrile, hemodynamically stable with a hemoglobin level near her baseline (12 g/dL) and was discharged home with analgesics and encouraging hydration. Five days later she presented with complaints of worsening severe abdominal pain, nausea, vomiting, fever and chills and was referred to the emergency department. On admission, she was noted to be febrile (102.3F), tachycardic (118 beats/min), tachypneic (28 breaths/min) with an oxygen saturation of 100% on room air. Her physical exam was notable for labored breathing. The patient retested positive for SARS-CoV-2 by nasopharyngeal PCR. Laboratory results on admission were notable for low white cell count at 2.9 x 103/µl with absolute lymphocyte count of 360, hemoglobin of 11 gm/dl and elevated inflammatory markers (ferritin 4500 ng/ml, lactate dehydrogenase 1500 U/L, C-reactive protein 25 mg/L, procalcitonin 7.4 ng/mL, fibrinogen >700 and D-dimer 6000 mg/dl). CT abdomen/pelvis imaging showed enlarged spleen at 24 cm as well as splenic hypodensity measuring 8.6 x 0.7 cm, consistent with splenic infarct with surrounding peri-splenic edema and presence of bilateral patchy consolidation in the lungs suggesting viral pneumonia. She was started on therapeutic enoxaparin at 1mg/kg twice daily and hydroxychloroquine (600 mg every 12 hours) for COVID-19 pneumonia in addition to supportive care. The following day patient had waxing and waning mental status which prompted evaluation with MRI of head revealing a small acute infarct in the left temporoparietal periventricular white matter and medial right parietal cortical infarct. Differential diagnosis at this time included hypercoagulable state from COVID-19, embolic shower or catastrophic anti-phospholipid syndrome (APS). Vaso-occlusive crisis was thought to be a possibility as well but was low on differential given the patient’s reported history of “sickle cell trait”. APS panel and inherited hypercoagulable panel including factor V Leiden, prothrombin gene mutation in addition to JAK2, BCR-ABL and HB electrophoresis was sent. On day 3 of admission, her hemoglobin dropped to 4.8 gm/dl although there were no overt signs of hematoma or bleeding. The patient required rapid escalation of care and was transferred to the intensive care unit in which code fusion was initiated and a total of four units of packed red blood cells were transfused. Anticoagulation was held given the acute drop in hemoglobin to prevent further bleeding complications. Urgent CT abdomen and pelvis angiography did not reveal any evidence of bleeding but showed increase in size of the splenic infarct measuring 14 x 2 x 9 cm. Her laboratory results prior to transfusion showed haptoglobin less than 20 mg/dl, total bilirubin 2.1 mg/dl, reticulocyte count of 2% suggestive of hemolysis. Repeat hemoglobin level post transfusion was 10.4 gm/dl. Overnight, patient became more hypoxic with increased oxygen requirements requiring rapid sequence intubation using COVID-19 airway precautions. She also received a dose of tocilizumab for cytokine storm and was also treated with cefepime for bacterial superinfection. On Day 4, the results of hemoglobin electrophoresis prior to transfusion showed HbA-0%, HbS-50.5%, HbC-45.2%, confirming HbSC disease. The patient’s hemoglobin stayed persistently above 10 gm/dl twenty-four hours post transfusion, so exchange transfusion was not attempted. Her post transfusion hemoglobin electrophoresis showed HbA 53%, HbS 20%, HBC 22%. Day 5, patient was extubated and did not have any focal neurological deficits. Other pertinent negative laboratory testing included antiphospholipid panel, JAK 2, BCR- ABL, Prothrombin gene and Factor V Leiden mutations. Given the extent of infarct and possibility of hypercoagulable state from COVID 19, anticoagulation was resumed with close laboratory monitoring. Patient was eventually discharged on day 8 of hospitalization without any abdominal pain, oxygen requirements and residual neurological deficits (Figure 1).
Hemoglobin SC disease is expected to have a milder clinical course compared to homozygous SCA. However, because of the underreported cases, clinical diagnosis is often delayed because of the suspected benign course and milder hemolytic anemia. The mechanism by which HbSC leads to cell dehydration is by enhanced potassium chloride cotransport. The loss of potassium results in RBC dehydration which leads to polymerization, sickling resulting in a prothrombotic state [6]. HBSC as a spectrum is associated with increased circulating tissue factor procoagulant factor, increased markers of thrombin generation, decreased levels of natural anticoagulant proteins, and evidence of platelet and fibrinolytic system activation making patients prone to thrombotic complications [7]. In addition, the altered morphology of sickle red blood cells (RBC) may also alter the properties and dynamics of clot formation. The hemostatic perturbations among persons with HbSC are not as pronounced but, nonetheless, are apparent. For example, laboratory markers of coagulation activation such as TAT complexes, d-dimers, and prothrombin fragment are elevated in persons with Hb SC compared with those with Hb AA [7]. This was evident in our patient as her inflammatory markers were above baseline levels. The outbreak of coronavirus disease 2019 (COVID-19) which began in Wuhan, China, in late 2019 resulted in over 219 million positive cases, and 4.5 million deaths worldwide [8]. Risk factors for adverse outcomes in Covid 19 patients include advanced age, comorbidities (e.g., obesity, diabetes, cardiovascular, pulmonological and renal diseases) but also people with weakened immune systems from a medical condition or treatment are at a higher risk [9]. Individuals with sickle cell disease (SCD) are a vulnerable group of patients, with a higher risk of severe complications of COVID 19 than the general population. The spectrum of severity of COVID-19 infection could range from being asymptomatic, mild infection to severe disease with respiratory failure, shock, stroke, thrombotic complications, and multi-organ failure leading to death. The pathogenesis of hypercoagulability in COVID-19 is ill-defined but a few proposed pathogenesis of hypercoagulability in COVID-19 consist of all three components of Virchow’s triad, including endothelial injury, stasis, and hypercoagulable state [10]. Increased cytokines are released, such as interleukin (IL)-6, and various acute-phase reactants in COVID-19 and also lead to endothelial injury [11]. Reports also suggest activation of alternate and lectin complement pathways (C5b-9 [membrane attack complex], C4d, and mannose-binding protein-associated serine protease 2 [MASP2]), leading to further endothelial cell injury [12]. A hypercoagulable state is seen due to several coagulation abnormalities from elevated circulating prothrombotic factors such as elevated von Willebrand factor (vWF), factor VIII, D-dimer, fibrinogen, neutrophil extracellular traps, prothrombotic microparticles, and anionic phospholipids [13]. Elevated levels of D-dimer and fibrinogen have been observed to correlate with illness severity and 28-day mortality [14]. Recent studies have shown higher case fatality rate in patients with sickle cell disease of 10.9% compared to 3.3% in general population in which bout 75% of patients with SCD required hospitalizations secondary to COVID-19 [15]. Pain was generally the most common presentation followed by acute chest syndrome, fever, tachycardia, hypoxia however splenic sequestration was not reported in this recent study of SCA and COVID-19 [15]. Since COVID 19 and HBSC both result in a hypercoagulable state this can precipitate to cause worsening clinical outcomes in patients with HbSC who have been asymptomatic prior to this. Our patient with HSBC hemoglobinopathy with no prior hospitalizations and relatively asymptomatic life course prior to this event likely had a worsening hospitalization course after being infected with COVID-19. She had severe vaso-occlusive crises leading to multiple infarcts, splenic sequestration crises resulting in an acute drop in hemoglobin and splenic infarcts which was incited with her medical history of hemoglobinopathy and infection with COVID-19. However, her reticulocyte count was not significantly elevated which is probably due to bone marrow suppression from the COVID-19 infection itself and probably due to HBSC having milder hemolysis compared to SCA. She did not require any exchange transfusion during her stay as her HbS level dropped to 20% post packed red blood cell transfusion. Splenic sequestration crisis is a potentially fatal complication of sickle cell disease with mortality of 10-15%. In HbSS disease, it occurs at a mean age of 4 years, could range from 2 months of age to 14 years [16]. However, in the milder variant like HbSC disease it could occur later in life as they have persistent splenic function which can lead to splenic infarctions and sequestrations5. Inciting factors for acute splenic sequestration in adults are not well defined. In our patient the inciting factor can be due to COVID-19 given known course of thrombotic complications which never occurred prior to this infection. Risk of recurrent splenic sequestration is high after the first episode especially in children and splenectomy is advocated usually after the first major attack [17]. However, the risk of recurrence in adult patients is unclear and specific guidelines for splenectomy is not outlined.
Special attention in hemoglobinopathy patients with COVID-19 should include venous thromboembolism prophylaxis, conservative fluid management and avoidance of hypoxemia to decrease the chances of worsening vaso-occlusive consequences. Notably, this young adult was advised for conservative management at home with his initial primary care physician visit. The patient remained in close contact with his primary care physician, and this allowed a rapid referral to the ED in the setting of worsening symptoms. Dedicated patient pathways and clinical protocols are vital for patients with hemoglobinopathies diagnosed with Sars-Cov-19.
As the pandemic continues to evolve with the novel COVID-19 virus, many questions are brought upon in patients with history of a hemoglobinopathy concerning for course of presentation, complications, management and prognosis. Of those requiring admission to the intensive care unit many carry a comorbidity including hematologic comorbidities although hematologic conditions may not be an independent risk factor of severe COVID-19. This case suggests that patients with even milder forms like hemoglobin SC disease could have life threatening complications from the novel coronavirus infection. It is also important to educate the patients that HbSC disease is not the same as sickle cell trait as providing accurate history during the initial encounter will aid in early diagnosis and prevention of potential complications. The concurrence of a hemoglobinopathy and COVID-19 may warrant heightened clinical suspicion for unusual outcomes. Hence, providers must remain vigilant while treating any patient with any hemoglobinopathy in the setting of COVID-19.
From the patient as there is no patient identifiable data included in this case report.

![]()

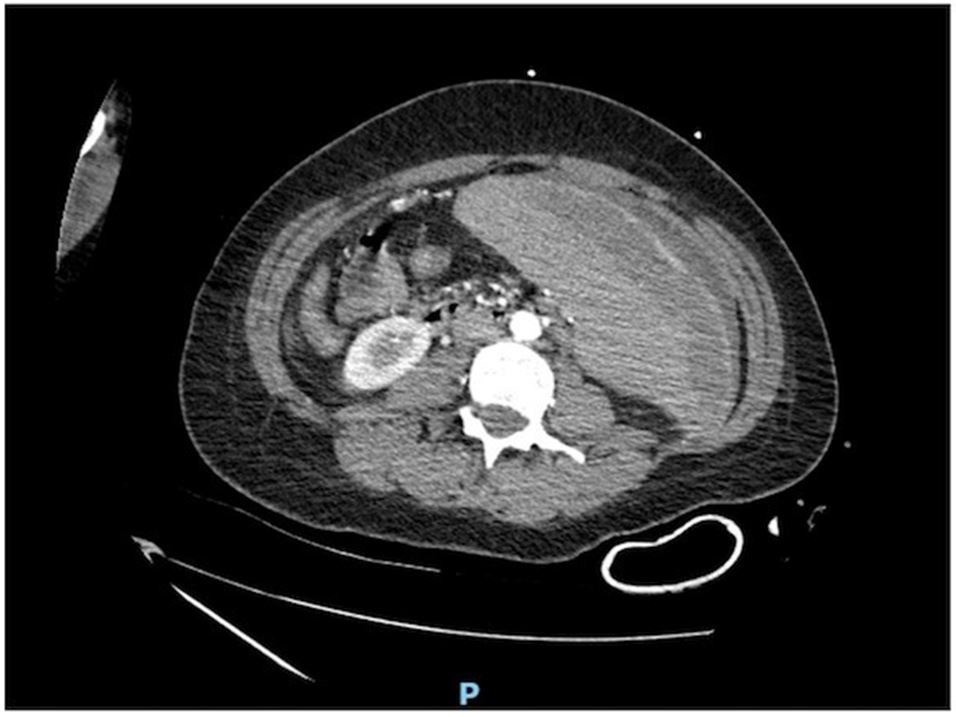 |
| Figure 1: Splenomegaly measuring 24cm in size. Linear splenic hypodensities largest measuring 14 X 2 X 9 cm with surrounding perisplenic edema. Findings consistent with splenic infarcts |




































