Open Access
Case Report
Max Screen
Copyright: © 2025 Dedeoğlu O. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Primary coenzyme Q10 deficiency-4 (CoQ10D4) is an autosomal recessive disorder characterized by childhood-onset of cerebellar ataxia and exercise intolerance. Molecular pathology responsible for clinical findings is mitochondrial respiratory chain dysfunction. The main clinical manifestation involves early-onset exercise intolerance, progressive cerebellar ataxia and movement disorders. Some affected individuals develop seizures and have mild mental impairment, indicating variable severity. Coenzyme Q8A (CoQ8A) gene mutations are responsible for this disease. Here we present a patient with tremor and cerebellar atrophy in which we detected a new mutation in the CoQ8A gene. The patient's findings were compatible with juvenile-onset CoQ10D4. Therefore, we reviewed the clinical spesifities of 11 juvenile-onset CoQ10D4 patients reported to date, as well as the patient's presentation.
Keywords: Primary Coenzyme Q10 Deficiency; CoQ8A, Novel Mutation; Cerebellar Atrophy; Spinocerebellar Ataxia; Mitochondrial Respiratory Chain Dysfunction
Primary coenzyme Q10 deficiency-4 (CoQ10D4), also known as autosomal recessive spinocerebellar ataxia-9 (SCAR9), is an autosomal recessive disorder with mitochondrial respiratory chain dysfunction. The main clinical manifestations of CoQ10D4 involve exercise intolerance, progressive cerebellar ataxia and movement disorders. Some affected individuals develop seizures and have as a mild mental impairment, indicating variable severity. It can also be presented with mitochondrial myopathy, hypogonadism and steroid‐resistant nephrotic syndrome [1,2]. The age of onset of the disease varies from infancy to late adulthood. CoQ10D4 is caused by homozygous or compound heterozygous mutation in the CoQ8A gene (ADCK3 or CABC1). The CoQ8A gene encodes a protein, the homologue of the yeast CoQ8 gene, which is involved in the Coenzyme Q10 (CoQ10) biosynthesis pathway. CoQ10 is essential for as the proper functioning of the mitochondrial respiratory chain. CoQ10 acts as an electron carrier in the mitochondrial respiratory chain and plays a role as an antioxidant and membrane stabilizer. These two functions constitute the basis for supporting the clinical indication of CoQ10 [3,4].
In this study we report a 15- year old female patient with hand tremor, prominent handwriting impairment who was born to healthy consanguineous parents. Brain Magnetic Resonance Imaging (MRI) showed significant cerebellar atrophy. Whole exome sequencing revealed the presence of a novel mutation (p. I341T c.1022T> C) in the CoQ8A gene. Clinical, laboratory and treatment response of juvenile-onset CoQ10D4 patients reported to date are also evaluated in this article.
Our patient (pedigree showed in Figure 1) is a 15-year-old female who was born to healthy consanguineous parents and had one asymptomatic sibling. She was born after an uncomplicated pregnancy and delivery at term. She sat, rolled and crawled at appropriate ages. In her 10 years old she suffered from tremor in her hands. She started to experience awkwardness at 13 years of age and also complained of writing difficulty (Video 1). Her past medical history is significant for successfully treated childhood febrile convulsion. Her uncle and cousin has similar complaints. She is not dysmorphic, but does have epicanthus. Her eye movements are normal and she has no visual disturbance or nystagmus. Neurological examination revealed mild shakiness, bilateral dysmetria, intention tremor on nose-finger and heel-shin tests and inability to walk in tandem. Motor and sensory examination and deep tendon reflexes were normal. In serum, cholesterol, lactate and creatine kinase and thyroid antibodies were normal. The remaining blood tests, including liver function and plasma levels of vitamins (B12, A, D, E) were all normal. Electromyography revealed mild myopathy in the arms. No cardiac hypertrophy or fundus abnormalities were noted hearing were normal also. Brain MRI showed prominent cerebellar sulci with a loss of height of the cerebellar hemispheres and atrophy of the vermis (Figure 2). The Wechsler intelligence test was normal. The consanguineous parents (first-degree cousins) showed no neurologic abnormalities. Scale for the assessment and rating of ataxia (SARA) score was 9 (gait 2.0, stance 1.0, sitting 0, speech 0, finger chase 2.0, nose-finger test 2.0, fast alternating hand movements 2.0, and heel-shin slide 1.0). The result of 12-step stair test was normal with 10.6 sn. Whole exome sequencing (WES) examination was performed because of cerebellar atrophy, ataxia, positive family history and parental consanguinity. WES revealed the presence of a novel mutation (p.I341T c.1022T>C) in the CoQ8A (ADCK3, NM_020247.5) gene, homozygous. The clinical significance of this variant was not reported in the ClinVar and Human Gene Mutation databases (HGMD). The clinical significance of this variant was not reported in the ClinVar and HGMD databases. The VarSome modeling program predicted this variant as likely pathogenic. In the family study, it was shown that the mother, father and brother of the patient carried the same variant as heterozygous (Figure 3). Homozygous or compound heterozygous mutations of the CoQ8A gene cause primary Co Q10 deficiency-4 (OMIM #612016). Primary CoQ10D4 is an autosomal recessive disorder characterized by childhood-onset cerebellar ataxia and exercise intolerance. The patient was diagnosed with CoQ10D4 because the family study results and the detected variant overlapped with clinical findings. CoQ10 supplementation was immediately started with ubidecarenone of 40 mg three times per day. After 2 weeks of therapy, her tremor notably improved. Her SARA total score improved from 9 to 5.
Mutations in CoQ8A gene can result in primary CoQ10 deficiency type 4 that patients typically present with clinical features including ataxia or a more subtle gait instability. Other neurological abnormalities have been reported include adolescence onset exercise intolerance due to fatigability, seizures, stroke-like episodes, intellectual disability, spasticity, ophthalmic involvement, decreased visual acuity, sensorineural hearing loss, depression [5,6].
Juvenile-onset was characterized by a variable combination of writing difficulties clumsiness and tremor with ataxia, unsteady gait, myoclonus or dystonia. Clinical severity is reported juvenile-onset (11–18 years) was classified mild (slow progression, less severe symptoms and absence of cognitive impairment) to severe (presence of severe epilepsy and/or regression, association with poor cognitive and/or motor outcome such that subjects are reliant upon caretakers for all activities of daily living). The clinical, laboratory and treatment response characteristics of juvenile-onset CoQ10D4 patients reported to date are summarized in Table 1.
Our patient showed a less severe course and as in literature cerebellar symptoms were mild, writing difficulties were obvious. Although the origin of hand clumsiness may in part be cerebellar, particularly when associated with gait ataxia, the dystonic nature of hand clumsiness and neck tremor has been demonstrated in CoQ10D4 patients through electromyographic recordings CoQ10 levels are reduced in skeletal muscle and less frequently in fibroblasts, but their correlation with disease severity is still debated [7]. It has been reported that there was no difference in patterns of clinical presentation between subjects with missense and nonsense mutations or for mutations distributed throughout specific regions of the protein [8]. Cognitive impairment is often observed in primary Co Q10 deficiency cases with epileptic encephalopathy. Our patient exhibited a relative normal cognitive state. The absence of seizures, exercise intolerance or ocular dismotility confirms the heterogeneity of this disorder and the lack of correlation between CoQ10 residual levels and disease severity. This case emphasizes the importance of an early molecular diagnosis for suspected inherited ataxias, particularly given the availability of approved treatments for some subtypes similar to our patient.
The additional compelling evidence for the CoQ10 deficiency in our patient comes from clinical responses to CoQ10 supplementation, including the improvement in exercise intolerance and unsteady gait, a response similar to that found in the majority of cases of primary CoQ10 deficiency [9,10]. Dosage and course of CoQ10 supplement have not been standardized and results have been variable. The dose of oral CoQ10 (ubiquinone, ubiquinol, idebenone and ubidecarenone) ranged from 5 mg/kg/day to 3000 mg/day in the treatment of CoQ10 deficiencies. Most CoQ10D4 patients experienced symptomatic improvement [11,12]. Early and sustained CoQ10 supplementation appears to be important for a favorable outcome, suggesting that persistent ongoing damage to target tissues and irreversibility of established damages are determinants of therapeutic efficacy [8,13].
We administered oral ubidecarenone 100 mg, two times a day for 2 weeks to our patient, with an initial subjective improvement of fatigue followed by a remarkable improvement of tremor with a substantially lower (by five points) total SARA score. Regarding previous reported juvenile-onset cases, ubiquinone therapy did not lead to significant improvement of the neurological status in most patients [10,14,15]. Severe forms seem to be less responsive to CoQ10 supplementation while patients with ataxia tend to have a better response. CoQ10 is frequently reduced in the muscle of patients with mitochondrial myopathy. It is unfortunate that the patient declined muscle biopsy since the High-performance liquid chromatography (HPLC) assay for the CoQ10 level in skeletal muscle is the golden standard for CoQ10 deficiency.
Our aim is to highlight the clinical and genetic heterogeneity of CoQ10 deficiency and also to report a novel mutation of the CoQ8A gene that has not been previously reported in the literature. Gait ataxia, suggesting that cerebellar syndrome may be the common feature of juvenile-onset patients. The atypical presentation with prominent writing deterioration, possibly representing the initial manifestation of cerebellar disease, is an example of the extreme phenotypic variability of CoQ10 deficiency. Informed consent was obtained from the patient and parents.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Özge DEDEOĞLU, Ajlan TUKUN and Yahya LALELİ. The first draft of the manuscript was written by Özge DEDEOĞLU and Ajlan TUKUN. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.

![]()

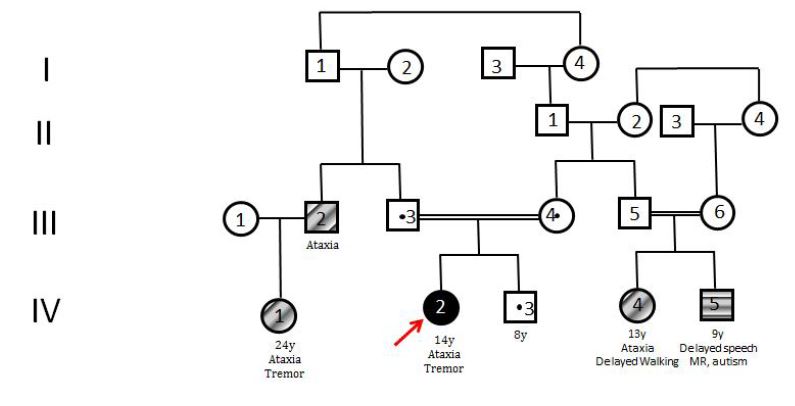 |
| Figure 1: Pedigree of the patient |
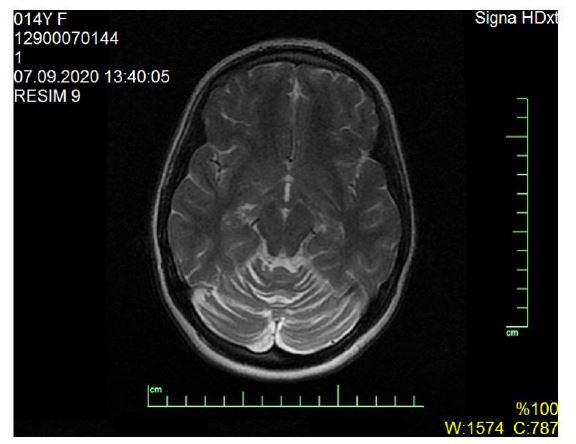 |
| Figure 2: Magnetic Resonance Imaging (MRI) showed prominent cerebellar sulci with a loss of height of the cerebellar hemispheres and atrophy of the vermis |
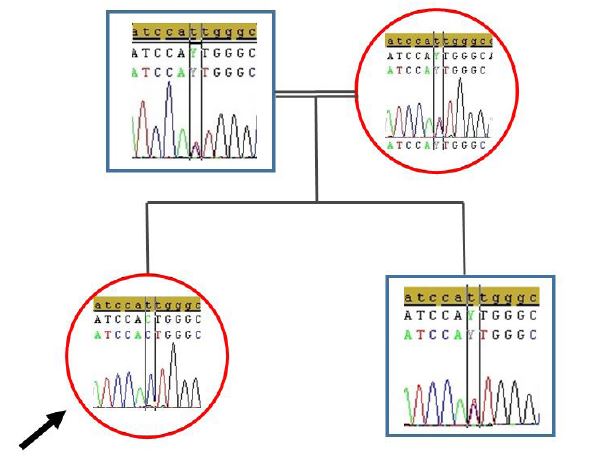 |
| Figure 3: Sanger sequence images of father (A); mother (B); patient (C); and brother (D); for the c.1022T>C mutation in the COQ8A gene |
Gender |
Onset |
Presentation symptoms |
Severity |
Clinical features |
MRI |
Variants |
Serum COQ10 |
Response |
References |
F |
15 | handwriting deterioration clumsiness | mild | gait disturbance , disartri,dysphagia tremor | cerebellar atrophy | c.811C > T (p.Arg271Cys) c.911G > A (pAla304Thr) | normal | no response | Howarth, 2012 |
F |
15 | ataxia | severe | Intractable seizures, DA, global DD, strabismus | vermis atrophy, T2 signal alterations in cerebellum and vermis | c.1334_1335del (p.Thr445Argfs*52 | unknown | unknown | Sun, |
F |
10 | slurred speech and unsteady gait | severe | difficulty in writing and holding objects | cerebellar atrophy | c.1844_1845insG; p. Ser616Leufs*114 | normal | marked clinical improvement | Liu, |
F |
15 | ataxia | severe | Intractable seizures, DA, global DD, strabismus | vermis atrophy, T2 signal alterations in cerebellum and vermis | c.1334_1335del (p.Thr445Argfs*52 | unknown | unknown | Sun, |
F |
10 | slurred speech and unsteady gait | severe | difficulty in writing and holding objects | cerebellar atrophy | c.1844_1845insG; p. Ser616Leufs*114 | normal | marked clinical improvement | Liu, |
M |
14 | tremor and unsteady gait | moderate | myoclonus |
| unknown | improvement in speech and fatigue | Liu, |
|
M |
11 | focal dystonia, writing difficulties | mild | ataxia disartri tremor | normal | c901C > T (p.Arg301Trp) c.1399–3_1408del | unknown | marked clinical improvement | Chang, |
F |
12 | unsteady gait, falls, and upper extremities tremor | mild | dysarthria, gait ataxia | cerebellar atrophy | (c.902G>A, p.Arg301Gln and c.1844_1845insG, p.Ser616Leufs*114) | unknown | remarkable improvement in ataxia | Chang, |
M |
9 | exercise intolerance | moderate | unsteady walking, with involuntary jerking and tremor of the head | cerebellar atrophy | c.1334_1335del (p.Thr445Argfs*52 | decreased | fatigue and exercise intolerance notably improved. | Zhang, |
F |
10 | mild dysfluent speech and clumsiness | mild | mild dysarthria | cerebellar atrophy | p. Thr584delACC (c.1750_1752delACC). | unknown | partial response | Blumkin, |
F |
9 | progressive speech and coordination difficulties | mild | reduced exercise intolerance | cerebellar atrophy | e c.1042C > T mutation in the CABC1 gene | unknown | unknown | Gerards, 2010 |
M |
20 | gait imbalance | moderate | frequent falling and dysarthria | cerebellar vermis atrophy | (c1511_1512delCT) | normal | clinical improvement | Barca, |
F |
15 | Writing | mild | mild shakiness, bilateral dysmetria, intention tremor, inability to walk in tandem | cerebellar atrophy | c.1022T>C | unknown | clinical improvement | Present case |




































