Open Access
Research Article
Max Screen
Copyright: © 2025 Zheng H. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Developmental delay (DD) / intellectual disability (ID) is considered one of the most genetically heterogeneous human diseases. Herein, we described a Chinese boy affected by DD/ID with 3-methylcrotonyl-CoA carboxylase deficiency (MCCD) and 3-hydroxy-3-methylglutaric aciduria (3-HMG), which associated with a novel missense hemizygous mutation, c.4442C>T, within the acidic domain of HCFC1 gene identified by whole exon sequencing (WES) and validated by Sanger sequencing. Pedigree analysis showed that the mutation was inherited from the proband’s mother and grandmother. Molecular functional analysis showed that HCFC1 c.4442C>T mutation increased the mRNA and protein expression in HET293T cells. Simultaneously, the mutation affected subcellular localization and interrupted the nuclear degradation of HCFC1 in the cells. In addition, the mutation decreased the expressions of MCCC1, MCCC2, HMGCL, relating to MCCD and 3-HMG. Moreover, HCFC1 c.4442C>T inhibit cell apoptosis. This X-linked disease revealed a new insight into a distinct disease mechanism that transcriptional dysregulation can lead to metabolic disorders with a much more serious and complex clinical phenotype. This novel mutation, related to DD/ID with MCCD and 3-HMG, was the first reported genetic mutation occurring within the acidic domain of HCFC1 to date. The results extend the mutation spectrum of the HCFC1 gene and reveal a new insight into a distinct disease mechanism of the mutated HCFC1 gene.
Keywords: Developmental delay; Intellectual disability; HCFC1
List of abbreviations: DD: Developmental Delay; ID: Intellectual Disability; MCCD: 3-Methylcrotonyl-CoA Carboxylase Deficiency; 3-HMG: 3-Hydroxy-3-Methylglutaric Aciduria; WES: Whole Exon Sequencing
Developmental delay (DD) / intellectual disability (ID) is characterized by impaired cognitive abilities and limited ability to adapt to the natural and social environment, mostly with intelligent scores equal or less than 70 [1]. ID, also known as mental retardation, is often used to describe people who are over 7 years of age with reliable and stable IQ, while DD refers to infants and preschool children who are under 5 years of age [2]. DD/ID has become one of the biggest social and medical challenges, affecting 2%-3% of the population [1-3]. Genetic factors, including both chromosomal aberrations and single gene mutations, in addition to environmental factors are contributed to DD/ID, which was considered one of the most genetically heterogeneous human diseases [1-3]. Over 100 genes have been reported associated with X-chromosome-linked intellectual disability (XLID), and HCFC1 was among them [4,5].
HCFC1 (OMIM 300019), the Host Cell Factor C1 gene, locates at Xq28 with the expression in mitochondria and nucleoplasm. It does not encode any enzyme, but it is a predicted transcriptional co-regulator related to cell proliferation and mitochondrial biogenesis [6-8] and is involved in the regulation of over a quarter of human promoters [9]. Presently several mutations in HCFC1 have been found as the likely cause of mild non-syndromic ID [4]. Furthermore, the high expression of HCFC1 was demonstrated to play an important role in brain development of a murine model [10]. Mutations and abnormal expression can affect the differentiation of neural progenitor cells, the growth of hippocampal neurons, the degree of neurite arborization and cause high amounts of neuronal death [11-14].
Here, we investigated a Chinese 2-year-old boy with DD and found a novel hemizygous mutation c.4442C>T (rs199798029) in HCFC1. After studying the function and potential pathogenic mechanism of the mutant gene, we drew a conclusion that the novel mutation of HCFC1 ultimately caused DD accompanied by 3-methylcrotonyl-CoA carboxylase deficiency (MCCD) and 3-hydroxy-3-methyl glutaric aciduria (3-HMG). Our findings will shed further light into providing a reference model for studying the potential pathogenic mechanism of DD/ID caused by variations of HCFC1.
The proband (Ⅲ-1), a 2-year-old boy, was referred to the Department of Neurology of Nanfang Hospital because of DD. The patient’s medical records were collected by a skilled neurologist. More than 200 normal male controls, matched by ethnic origin with unknown biochemical or neurological phenotypes, were recruited from healthy individuals. In accordance with the ethical committee standards of Nanfang Hospital, patients and relatives analyzed in the study signed informed consent forms prior to inclusion in our study.
Peripheral venous blood was collected from all family members of the proband. Target capture and next-generation sequencing were performed by Keybay Co., Ltd and one significant mutation locating at exon 18 of HCFC1 was identified. Regions including this mutation were generated by polymerase chain reaction (PCR) using primers: F-ACAAGAGCGACATCTGGCGT and R-GTCCGTCCAGAGTCCACCAG, and verified using Sanger sequencing. 200 healthy male volunteers (from ages 2 to 60) were analyzed by high-resolution melting analysis (HRM) with the primers: F-ACCGGTGTGGACTGCGT and R-GTGCCATCACGACAACCGT.
Secondary structures of wild-type and mutant HCFC1 were predicted by SOPMA (Self-Optimized Prediction Method with Alignment), while three-dimensional structures were predicted by I-TASSER (Iterative Threading Assembly Refinement) and PHYRE2 (Protein Homology/ analogy Recognition Engine V 2.0). PolyPhen2 (Polymorphism Phenotyping v2) software was used to predict the harmfulness of mutant HCFC1 and analyze the conservation of HCFC1 across species. Functional effects of the mutant protein were estimated by SOSUI (SOSUI engine ver. 1.11).
The wild-type coding sequence of HCFC1 with HindIII and BamHI restriction sites was generated using Phanta Max Super-Fidelity DNA polymerase (Vazyme Biotech, Co., Ltd, Nanjing, China), and two pairs of primers used were F1-GAGACGGGGTGAGCGAGAAG, R1-GAAGTGCGAATGCTGGGACG, F2-CCCAAGCTTATGGCTTCGGCCGTGT, and R2-CGCGGATCCGCTAGTCAGCTTCCTC. Next, the PCR products were cloned into the pcDNA3.1-FLAG vector (Life Technologies, Carlsbad, CA, USA), and the pcDNA3.1-FLAG-HCFC1 was made after being transformed into DH5α bacterial strain competent cells (Vazyme Biotech, Co., Ltd, Nanjing, China) and extracted by AxyPrep Plasmid Miniprep Kit (Axygen Scientific Inc., CA, USA). We also constructed the mutant plasmid, pcDNA3.1-FLAG-mHCFC1 by site-directed mutagenesis with the QuikChange Multi Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) with the primers: F-GTCCTCCACACTGATGCGGGCTGTGACCAC and R-GTGGTCACAGCCCGCATCAGTGTGGAGGAC.
PcDNA3.1-FLAG, pcDNA3.1-FLAG-HCFC1 and pcDNA3.1-FLAG-mHCFC1 were transfected into human embryonic kidney (HEK) 293T cells using Lipofectamine 3000 (Invitrogen) in Opti-MEM (Invitrogen). 6-8 hours after transfection, cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) until they can be accessed for different purpose.
48 hours after transfection, cells were fixed with 4% paraformaldehyde (PFA), and blocked by phosphate-buffered saline (PBS) containing 0.2% Triton X-100 and 5% bovine serum albumin (BSA). After being incubated with monoclonal mouse anti-FLAG M2 antibody (1:500, Sigma-Aldridge, St. Louis, Mo., USA) and goat anti-mouse IgG (whole molecule)-FITC antibody (1:64, Sigma-Aldridge, St. Louis, Mo., USA), cells were counterstained with 4',6-diamidino-2-phenylindole (DAPI; Sigma-Aldridge, St. Louis, Mo., USA) and viewed using LSM 880 with Airyscan (Carl Zeiss, Jena, Germany).
24 or 48 hours after transfection, the expressions of wild-type and mutant HCFC1 were examined at mRNA and protein levels. All reactions were performed in triplicate.
Complementary DNA was synthesized by PrimeScript RT reagent Kit (TaKaRa Biotechnology, Co., Ltd, Dalian, China). SYBR Green (Promega Biotech, Co., Ltd, Beijing, China)-based relative quantitative RT-PCR was used to measure mRNA, and signal intensity from HCFC1 was normalized to GAPDH. Three-step real-time PCR analysis generated two standard curves, which was the Ct value plotted against the log of the input mRNA concentration at five five-fold serial dilutions, for linear quantitation of mRNA, with y=-3.399x+23.352 (R2=0.999) for HCFC1 mRNA and y=-3.27x+18.902 (R2=0.998) for GAPDH mRNA. The primers for HCFC1 and GAPDH were as follows: F-CCATCACTTACGGGGTCCTAC and R-CGTAGATCACCAGCTTGGACTT; F-GTGAAGGTCGGAGTCAACG and R-TGAGGTCAATGAAGGGGTC. Gene expression levels were calculated with the (2–ΔΔCT) method in the SYBR Green system, and results obtained from three reactions were used for calculating mean values and standard deviations.
Cells were harvested and homogenized in RIPA lysis buffer (Beyotime) containing protease inhibitor cocktail (Sigma), and extractable proteins were examined by SDS-PAGE with 8% gels. After blocking with 5% skim milk (Becton, Dickinson and Company), membranes were incubated with mouse anti-FLAG M2 antibody (1:2000, Sigma-Aldridge) or GAPDH (MC4) mouse monoclonal antibody (1:2000, Ray Antibody Biotech), all followed by HRP-goat anti-mouse IgG (1:2000, Santa Cruz) before being detected by Immobilon Western chemiluminescent HRP substrate (Millipore).
24 hours after transfection, we analyzed the expressions of MMACHC related to the metabolism of cobalamin, MCCC1 and MCCC2 to that of MCCD, together with HMGCL to that of 3-HMG at the mRNA level by real-time PCR. The primers were: F-TGTAGCTGGGGCTGCTTACT and R-ACACACCTGATATGCGCTGG for MMACHC; F-ACTGGAGTACGGCAAGGAGA and R-CTGACGAAGGCTGTACCTCA for MCCC1; F-CTGAAGAGCCTTTATTTCCTGCTG and R-TCTAGCAATGACCTCTCGGACA for MCCC2; F-AGGGGGCATCAGGAAACTTG and R-CTTCTGGAGATTCACACCCGT for HMGCL. These experiments were performed in triplicate.
Cells were transferred at an initial concentration of 2.0×104 cells/well and cultured in 96-well plates 24 hours after transfection. 12, 24, 36, 48 and 60 hours after transference, 100μl culture medium containing Cell Counting Kit-8 (CCK-8, DOJINDO, Japan, 10μl) were added and living cells were evaluated by microplate reader at an absorbance rate of 450 nm.
24, 36 and 48 hours after transfection, cells were digested with trypsin and harvested. Annexin V-FITC/PI apoptosis detection kit (KeyGEN BioTECH, China) and cell cycle kit (BestBio, China) were used respectively to check the apoptosis rate and the cell cycle through flow cytometry. The early apoptosis rate was taken by calculating the percentage of cells in the right lower quadrant, and the total apoptosis rate by both in the upper right and lower right quadrants.
The proband (III-1), a 2-year-old boy, was administered to our department because of DD, together with athetosis. Physical examination of the proband showed weakened deep tendon reflex, hypotonus (Class III), bilateral athetosis (+), bilateral Babinski (+), ATNR (+), TLR (±), and meningeal irritation (-), in addition to DD (Table 1). The proband was unable to crawl, to stand alone, or be supported by hand. Irritant tension, asymmetrical posture and negative grasp were observed, along with insensitive response, poor body axis cyclotron ability, poor sitting position balance, poor spinal load and control. Tandem mass spectrometry (Tandem MS) revealed decreased ratio of Met/Phe and Gly/Phe, in addition to the increased ratio of (C5DC/C6OH)/C8, (C5DC/C6OH)/C16, C4DC/C5OH and (C4DC/C5OH)/C8, which likely meant both MCCD and 3-HMG (Table 1). Furthermore, the proband’s brain magnetic resonance imaging (MRI) showed ventriculomegaly and dysplasia of the corpus callosum, in addition to the leukodystrophy (Figure 1).
The proband harbored a novel mutation from his mother and grandmother who presented heterozygous mutation, whereas both his mother and grandmother had neither clinical symptoms and signs, nor abnormal brain MR imagings. This mutation indicated an X-linked mutant gene which was relative to the DD phenotype with an X-linked recessive mode of inheritance (Table 1) (Figure 2A).
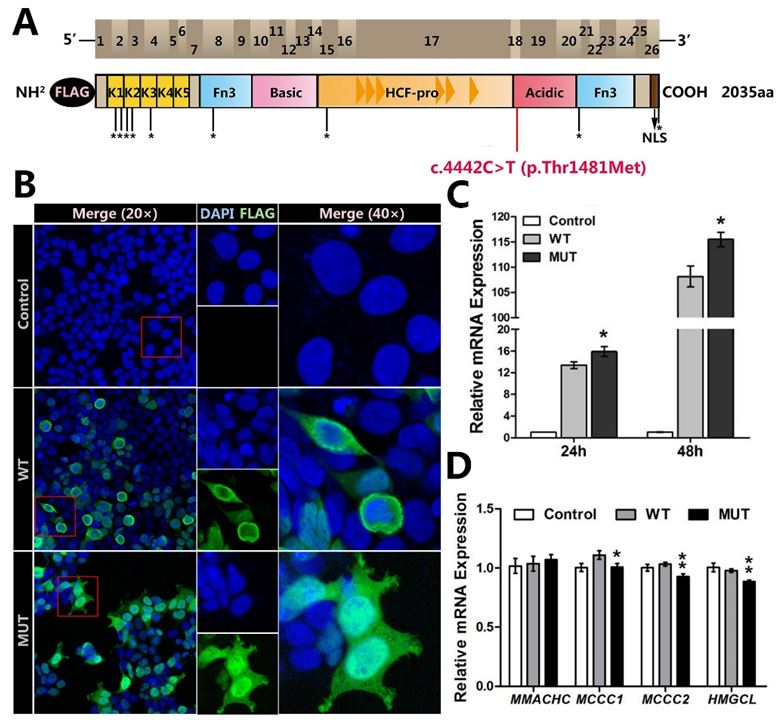
DNA sequencing revealed a missense mutation, c.4442C>T (rs199798029) within exon 18 of HCFC1 in the proband (Figure 2B). This mutation contributed to the mutant protein p.Thr1481Met, locating within the acidic domain of HCFC1 (Figure 3A), and it wasn't detected in 200 healthy male control samples (Figure 2C). The mutation was predicted to alter both the secondary structure and the three-dimensional structures of wild-type HCFC1 (Figure 2D). In addition, the bioinformatics analysis demonstrated that this variation was evolutionarily conserved across multiple species (Figure 2E) and the novel mutation was possibly damaging to humans.
After studying the subcellular localization, we found that akin to the localization of the wild-type HCFC1, the mutant HCFC1 was found not only enriched in the nucleus, but also in the cytoplasm. However, the mutant HCFC1 had more intranuclear accumulation than the wild-type one (Figure 3B), and it indicated that the c.4442C>T mutation may appear to affect subcellular localization and interrupt the nuclear degradation of HCFC1 in the cells, especially in the nuclei.
To further characterize the functional consequences of the HCFC1 mutation in vitro, we examined the expressions of HCFC1 by using qPCR and western blotting. The quantitation of the mutant HCFC1 was found higher than that of the wild-type at both mRNA (p<0.05) (Figure 3C) and protein level (p<0.01) (Figure 3F). Interestingly, two more bands, one around 130 kDa and the other 260 kDa, in addition to the expected 208 kDa were observed in the western blotting of both wild-type and mutant HCFC1 (Figure 3E) and these bands were probably the products associated with proteolysis.
Based on two results of Tandem MS, we hypothesized that the c.4442C>T mutation of HCFC1 may affect expression of other genes, which caused these two concurrent diseases, in the control of gene transcription. MCCD is due to mutations in MCCC1 and MCCC2, encoding for two subunits respectively that form the major rate-limiting enzyme, and leads to accumulation of 3-methylcrotonyl-CoA metabolites in blood and/or urine [12]. 3-HMG, also known as 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) lyase deficiency (HMGCLD), is attributed to mutations in HMGCL, and is characterized by nonketotic hypoglycemia, metabolic acidosis, and elevated urinary organic acids [13]. Given MMACHC is regulated by transcriptional complexes of HCFC1 [14], likewise, MCCC1, MCCC2 and HMGCL are supposed to be HCFC1 target genes, and the expressions of them are involved in the control of HCFC1 transcription.
As expected, the mutant HCFC1 caused the slightly reduced expression of MCCC1, MCCC2 and HMGCL (Figure 3D). However, no difference of MMACHC expression, which was contributed to X-linked Cobalamin type C-like deficiency (CblX) [10,14], was found between wild-type and mutant HCFC1 (Figure 3D).
As HCFC1 was previously reported to be of importance at multiple stages of the cell cycle [7,15,16], we investigated the biological effects of HCFC1 and its c.4442C>T mutant form on the proliferation, apoptosis and cell cycle progression of HEK293T cells. Evaluation of the apoptosis levels between control, wild-type and mutant HCFC1 indicated no effect was found on apoptosis in 24 hours after transfection (Figure 4A and B) (Figure S1). However, over-expression of mutant HCFC1 resulted in a significant decrease in both 36 and 48 hours after transfection. The total apoptosis rate of control, wild-type and mutant in 36 hours were 6.38±0.14, 6.39±0.35 (p<0.05), 5.52±0.47 (p<0.05), respectively. Then 48 hours later, mutant HCFC1 (4.66±0.32) resulted in a magnificent reduction of total apoptosis, compared with the control sample (6.08±0.11, p<0.01) and wild-type HCFC1 (7.23±0.11, p<0.001). These data suggested that HCFC1 c.4442C>T mutant would inhibit apoptosis, which was a modest yet significant loss of function, to some extent.
However, after 60 hours of growth, over-expression of wild-type HCFC1 caused an 11.23% increase (p<0.05) in growth, compared with control sample transfected with an empty pcDNA3.1-FLAG expression vector; while over-expression of mutant HCFC1 would not (p>0.05) (Figure 4C). In addition, no statistical difference was found in cell proliferation between control, wild-type and mutant HCFC1 (Figure 4D) (Figure S2).
Various studies have long demonstrated that both gain and loss of function mutations of HCFC1 can cause neurodevelopmental disorders such as DD/ID [5]. Here, we newly investigated the clinical findings of the proband carrying a novel hemizygous mutation in the HCFC1 causing DD finally. This mutation had not been previously reported and was predicted to be potentially damaging. The proband harbored the novel mutation from his mother and his maternal grandma, who did not exhibit any clinical symptoms. To explore the possible mechanism of this novel mutation, we studied the subcellular localization, mRNA level, protein expression, cell proliferation, apoptosis and cell cycle progression of this mutant HCFC1, in addition to its relationship to MMACHC, MCCC1, MCCC2, HMGCL. This c.4442C>T mutant we tested resulted in altered HCFC1 function consistent with a partial, but not complete effect. Other genetic factors such as pleiotropic genes might be further analyzed to elucidate the heterogeneity.
HCFC1 is known to interact with proteins involved in the regulation of multiple processes [7,17,18], including neural and neuronal progenitor cells of the developing brain [5]. HCFC1 has 26 exons, constituting several functional domains: the Kelch domain, the basic domain, HCFC1 proteolytic repeats domain (HCF-1PRO repeats), the acidic domain, nuclear localization sequence (NLS) and 2 fibronectin type 3 (Fn3) domains [5,14]. The HCF-1PRO repeats, consisting of six near-perfect functional repeats and two degenerate nonfunctional repeats, is where the proteolytic processing happened [19]. In the presence of proteolytic processing, HCFC1 is degraded into the fragments HCF-1N and HCF-1C [15]. As only the N terminal can be recognized, HCFC1 can be found as either a full-length protein, or fragments including the N terminus [5,20]. The 208 kDa blots indicated the full-length protein according to UniProt, and two more bands we observed, in addition to the expected 208 kDa in the western blotting of both wild-type and mutant HCFC1, probably meant fragment after proteolysis and its heterodimer. The blot around 130 kDa should be the HCF-1N, while the blot at around 260 kDa should be a heterodimer with non-covalent associations [21-23] and these heterodimers probably display independent functions [15,16]. The acidic domain is closed to the HCF-1PRO repeats. The result that both wild-type and mutant HCFC1 were found in the cell nucleus and cytoplasm demonstrated the main proteolytic process of the mutant HCFC1 has not been changed. However, the difference of the quantitation between cells with wild-type HCFC1 and cells with mutant HCFC1 found predominantly in the cell nucleus indicated that p.Thr1481Met mutant of HCFC1 might interrupt the proteolytic processing of HCFC1 in the nucleus to some extent rather than abolishing it completely.
Mutant HCFC1 inhibited apoptosis showed that this mutation has an effect on the development of HEK293T. Furthermore, overexpression of the HCFC1 in embryonic hippocampal neurons can result in a decrease in neurite growth, a reduction in the degree of neurite arborization, and increased neuronal death [4]. Following completion of our functional studies, the expression of the mutant HCFC1 was higher than that of the wild-type both in qPCR and immunoblot analysis. That may correspond to the proband clinical symptoms of DD, whereas the mechanism of this mutation needs further investigation.
It is credible that the Kelch domain is a protein-protein interaction motif and can bind to transcription factors and chromatin regulators [24]. Previous findings suggest the mutations of the Kelch domain of HCFC1, which is recruited to target promoter MMACHC by transcription factors [14,24], can lead to CblX featured by methylmalonic acidemia and hyperhomocysteinemia, in addition to ID [5,14]. Different from most cases caused by mutations of the Kelch domain of HCFC1, however, the proband in this case presented with DD with no evidence of methylmalonic acidemia and hyperhomocysteinemia. By studying the construction of HCFC1, we found the hemizygous mutation p.Thr1481Met located within the acidic domain, far away from the Kelch domain, and probably explained why the proband does not have obvious abnormal cobalamin metabolization.
Since there were no genetic mutations occurring in MCCC1, MCCC2 and HMGCL among the proband and his family, the mutant HCFC1 displayed negative effects on the expressions of these three genes providing evidence, which corresponds to the clinical symptoms including MCCD and 3-HMG. Both metabolic disorders can cause the injury to the central nervous system of children [25-27], thus we speculated MCCD and 3-HMG may further aggravate the symptom of DD. However, until now, the question of what actually induces HCFC1, as a co-regulator, to target and effect MCCC1, MCCC2, HMGCL in the transcriptional regulation is still unclear, and thus needs further study to be conclusive.
In conclusion, our data provide additional support for the involvement of HCFC1 in DD/ID. Together, we firstly identified a hemizygous mutation p.Thr1481Met of HCFC1 that was involved in DD/ID pathogenesis, accompanied by MCCD and 3-HMG. This is the first reported genetic mutation occurring within the acidic domain of HCFC1 so far, and it has been identified to the occurrence of DD/ID. This distinct disorder established the genetic heterogeneity for HCFC1, and induced a new insight into a distinct disease mechanism that transcriptional dysregulation can lead to metabolic disorders with a complex clinical phenotype. With additional research, the potential mechanism associated with DD/ID, and the contribution of p.Thr1481Met mutant to the pathology of the patient remains complicated and requires further investigation to be conclusive.
We thank the patients and their family members for consenting to this research. This work was supported by a grant from the National Natural Science Foundation of China (31970558), Natural Science Foundation of Guangdong Province (2018B030311033), Science and Technology Planning Project of Guangdong (2019A030317019), Research Grant of Guangdong Province Key Laboratory of Psychiatric Disorders (N201903) and the Foundation of President of Nanfang Hospital (2018B003).

![]()

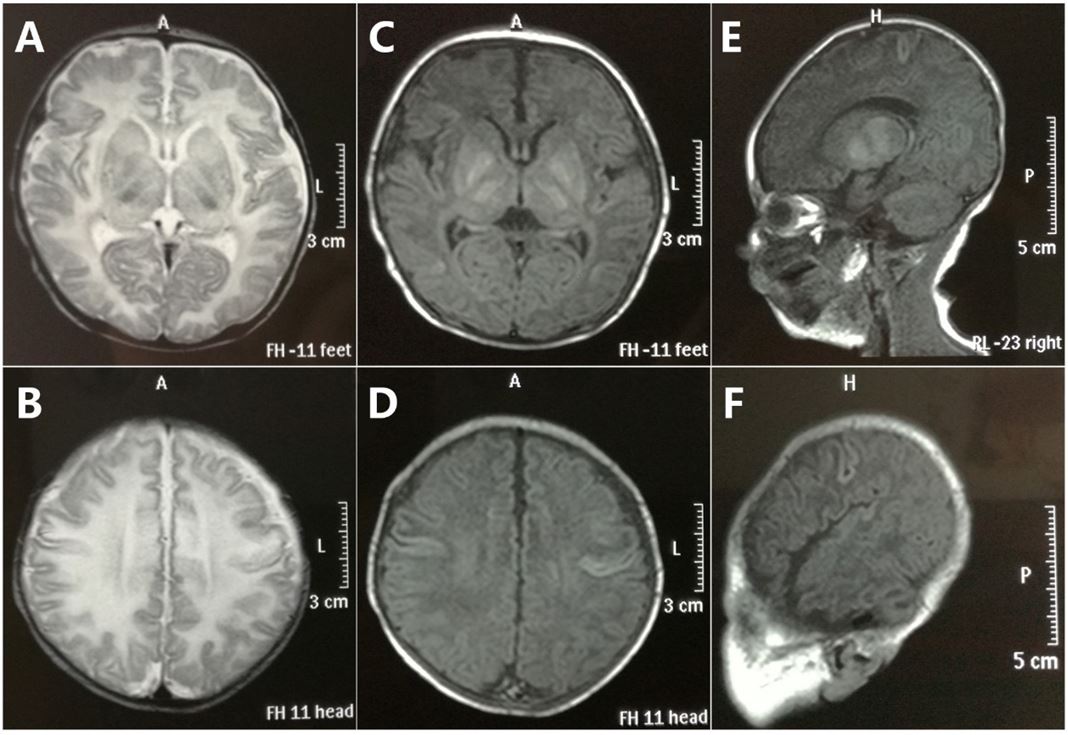 |
| Figure 1: MRI findings. Cross section T1 and T2 (A-D) cranial MRI of the proband showed serious leukodystrophy, while dysplasia of the corpus callosum was found through the T1 sagittal view (E, F) |
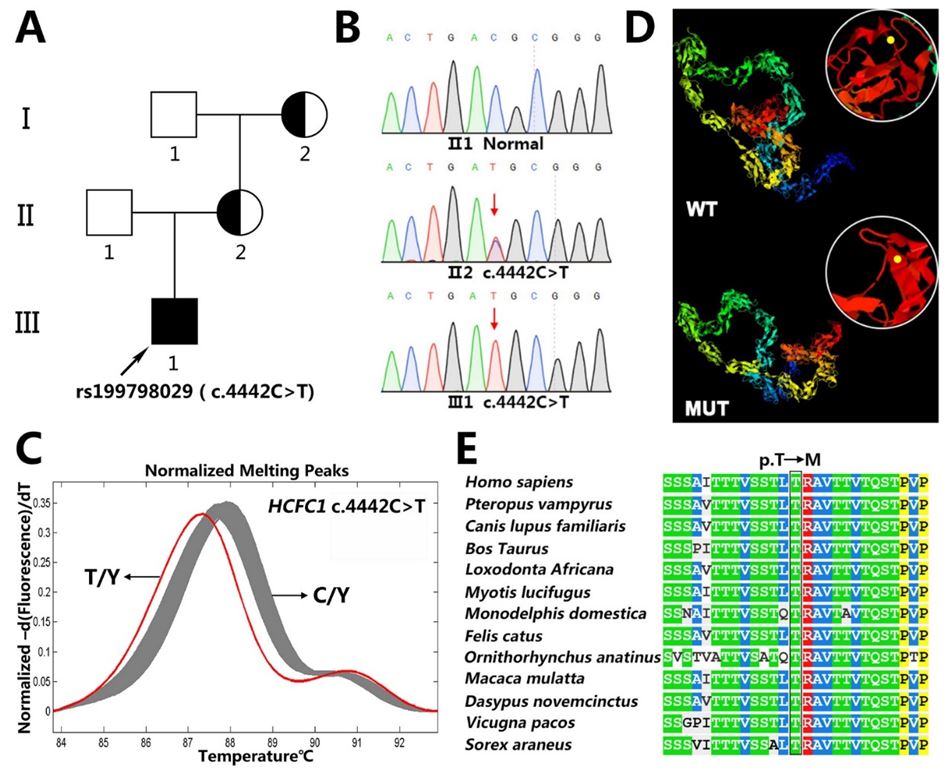 |
| Figure 2: Mutation screening and bioinformatics analysis: (A) Family pedigree. The arrow denoted the proband (Ⅲ-1); (B) Mutation screening. DNA sequences of wild-type (upper), heterozygote (middle) and mutant (lower) were validated by Sanger sequencing. The red arrow denoted the mutation; (C) High resolution melting analysis curves of proband’s (T/Y) and 200 healthy male volunteers’ (C/Y) genomic DNA at position rs199798029; (D) Prediction of wild-type (upper) and mutant (lower) protein structure by I-TASSER; (E) Sequence alignment of mutant amino acids across different species |
 |
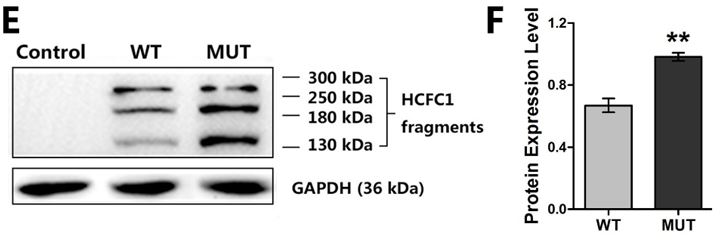 |
| Figure 3: Functional studies on the mutant HCFC1: (A) Known structural features of HCFC1 were illustrated in two linear scale cartoons. The top panel showed the 26 exons as brown boxes and the bottom panel indicated the predicted HCFC1 domains, including the Kelch domain K1-K5, the basic domain, HCFC1 proteolytic repeats domain (HCF-1PRO repeats), the acidic domain, nuclear localization sequence (NLS) and 2 fibronectin type (Fn3) domains. The location of mutant amino acid is marked with red, and locations of variants previously reported are marked with *[5,14]. Note also that the exogenously expressed HCFC1 was tagged by FLAG at the N-terminus; (B) HEK293T cells were transfected with empty vector (Control), wild-type (WT) or mutant HCFC1 (MUT) for subcellular localization studies. Immunofluorescent detection of exogenous wild-type and mutant protein used monoclonal mouse anti-FLAG antibody and fluorescent secondary antibody (green). Cell nuclei were counterstained with DAPI (blue); (C) Real-time PCR analysis of HCFC1 mRNA expression in 24 hours or 48 hours after transfection. Error bars represented the SEM of relative expression levels. *p<0.05, compared with wild-type; (D) Real-time PCR analysis of the mRNA expressions of MMACHC, MCCC1, MCCC2, HMGCL in 24 hours after transfection. *p<0.05, **p<0.01, compared with wild-type; (E,F) Immunoblot analysis of HCFC1. GAPDH was used as a loading control in all immunoblots. Right, the expression of a mutant plasmid was significantly increased compared with cells transfected with a wild-type plasmid. **p<0.01, compared with wild-type |
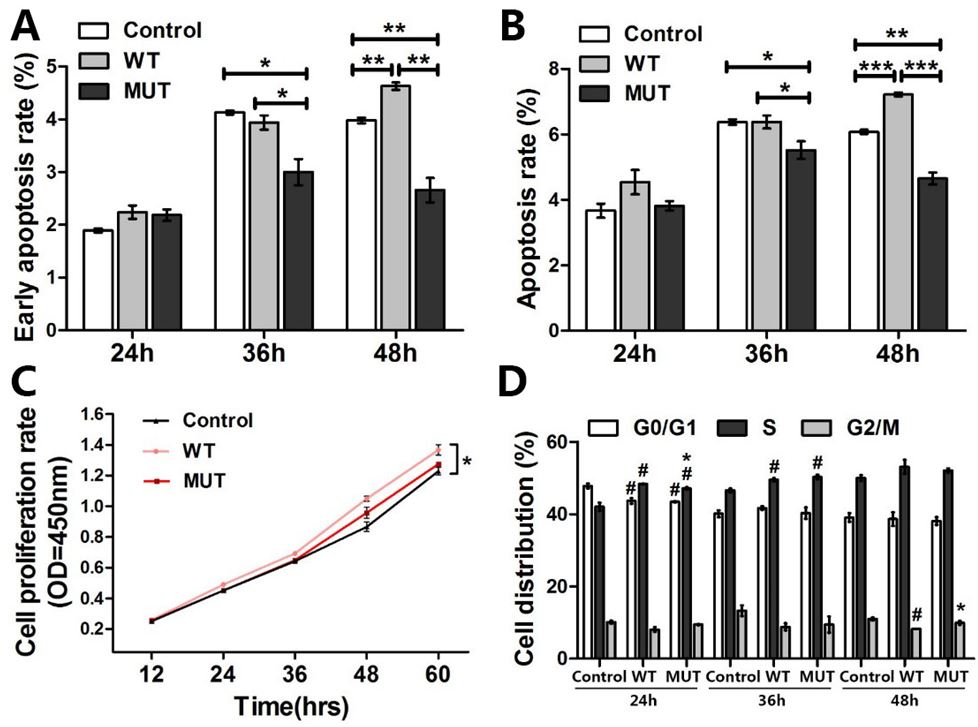 |
| Figure 4: Effects of p.Thr1481Met on cells proliferation, apoptosis and cell cycle progression: (A-B) Apoptosis levels of HEK293T cells were tested and cells are discriminated in early apoptosis (A) and total apoptosis (B) *p<0.05, **p<0.01, ***p<0.001, compared between each two groups; (C) The growth curve of HEK293T cells with CCK-8 assay. The average of triplicate plates with standard deviation indicated is represented; (D) Representative cell cycle distribution results are shown, and there were no statistical differences between wild-type and mutant HCFC1. *p<0.05, compared with wild-type; #p<0.05, compared with the control. These experiments were performed in triplicate |
 |
| Figure S1: Apoptosis levels of HEK293T cells were tested and cells are discriminated in early apoptosis (right lower quadrant) and total apoptosis (both right upper and right lower quadrants) through flow cytometry |
 |
| Figure S2: Representative cell cycle distribution results are shown through flow cytometry |
Subject |
HCFC1 |
Protein |
Age(y) |
Clinical presentation |
Tandem MS |
||||||||||||
ID/ |
ATNR |
TLR |
DTR |
Hypotonus |
Athetosis |
Babinski |
Meningeal irritation |
Met/ |
Gly/ |
C4DC/ |
(C4DC/ |
(C5DC/ |
(C5DC/ |
||||
I 1 |
No |
No |
U/M |
No |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
I 2 |
c.4442C>T |
p.T1481M |
U/F |
No |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
II 1 |
No |
No |
36/M |
No |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
II 2 |
c.4442C>T |
p.T1481M |
30/F |
No |
- |
- |
- |
- |
- |
- |
- |
↓ |
↓ |
N |
N |
↑ |
↑ |
III 1 |
c.4442C>T |
p.T1481M |
2/M |
Yes |
(+) |
(+) |
↓ |
Class III |
(+) |
(+) |
(-) |
↓ |
↓ |
↑ |
↑ |
↑ |
↑ |




































