Open Access
Case Report
Max Screen
ISSN: 2394-6520
Copyright: © 2021 Bronner MP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Background: Intramucosal lipomas are rare colonic polyps that pose diagnostic challenges, but they are important as potential clues to serious medical conditions. One third of intramucosal lipomas are reported to arise in Cowden syndrome, an inherited cancer syndrome. Until this report, no other known diseases or syndromic associations have been described for intramucosal lipomas. This is the first report of this rare colonic polyp in Li-Fraumeni syndrome, another rare but highly penetrant hereditary cancer syndrome. The intramucosal lipoma may therefore represent a new clue to this additional important inherited cancer syndrome
Case presentation: This 38-year-old man presented with a left temporal brain anaplastic astrocytoma. A striking family history of cancer (Figure 1) included a sister and mother with glioblastoma multiforme. The mother also had ductal carcinoma in situ of the breast. Genetic counseling and testing for hereditary cancer syndromes disclosed a pathogenic p.H193Q missense mutation in TP53, and the patient was diagnosed with Li-Fraumeni syndrome. The same TP53 variant was found in the patient’s mother, two brothers and three children. As part of Li-Fraumeni cancer screening, the index patient underwent upper and lower gastrointestinal endoscopy, revealing a single intramucosal lipoma measuring 4mm in size. No PTEN germline mutations were identified in the patient. This rare colonic polyp is unknown to occur in Li-Fraumeni syndrome and forms the basis of this report.
Conclusion : This is the first reported case of the rare colonic intramucosal lipoma in Li-Fraumeni syndrome and may represent a new colonic phenotype of this important cancer syndrome.
Keywords: Intramucosal lipoma; Li-Fraumeni Syndrome; Cowden Syndrome
Intramucosal lipomas are benign colonic polyps. They consist of mature adipose tissue within the colonic mucosal lamina propria between and surrounding colonic crypts (Figure 2A) [1]. This polyp is rare and unrelated to the relatively common submucosal lipoma of the colon. A very frequent endoscopic artifact, known as pseudolipomatosis coli, mimics intramucosal lipoma. It is caused by air insufflation into the colon during colonoscopy that under pressure enters into the colonic mucosa as gas bubbles that histologically simulate fat. Pathologists readily observe and ignore this mechanical artifact, which is present in up to half of colonic biopsies. Pathologists are not generally aware of the rare, true intramucosal lipoma; however, which is in the histologic differential of pseudolipomatosis coli. Accordingly, true intramucosal lipomas are likely under-recognized by pathologists [1]. Differentiation between a lipoma with true fat and the air bubble mimic of pseudolipomatosis is aided by both morphological features and S100 immunohistochemical staining (Figure 2) [1]. Morphologically, artifactual air bubbles are usually tiny and variable in appearance and are strongly associated with mucosal lymphoid aggregates (Figure 2B), whereas the rounded and characteristically much larger lipid vacuoles of true fat in intramucosal lipomas tends to be more uniform in shape and size and not typically associated with lymphoid follicles (Figure 2A). S100 immunohistochemical staining in true intramucosal lipomas reveals a distinctive although delicate and thin staining pattern of the outer rim of the adipocytes, which has been likened to an “eggshell” pattern (Figure 2C, arrows). Alternatively, air bubbles in pseudolipomatosis lack S100 staining altogether (Figure 2D, arrow) [1]. An important caveat concerning S100 staining is to avoid false positive over-interpretation of a true lipoma based
on the ever present S100-positive mucosal dendritic cells. Dendritic cells (Figure 2D, arrowheads) are scattered throughout the intestinal lamina propria and may be adjacent to or within air bubble collections of pseudolipomatosis (Figure 2D) [1]. Dendritic cells are small and have thin, spindled and delicate cytoplasmic processes that are also S-100 positive. They are distinctly different in appearance from the eggshell, full perimeter, S-100 pattern of true adipocytes.
Intramucosal lipomas are part of the multiple described phenotypes of Cowden syndrome, an autosomal dominant hamartoma and multi-organ cancer syndrome caused in part by mutations of the PTEN gene, which are identified in 25-35% of affected patients [2-6]. To date, no other genes have been identified. Patients with Cowden syndrome characteristically display skin lesions, most commonly multiple facial trichilemmomas, but also intestinal hamartomas, including colonic intramucosal lipomas. They harbor a high risk of multiple cancers, including breast, thyroid, endometrial, gastrointestinal, renal cell carcinoma, and melanoma [6-8]. These benign colonic mucosal lipomas have no known association with colonic adenocarcinoma in Cowden’s patients or as sporadic lesions and do not appear to be a cancer precursor. The clinical diagnosis of Cowden syndrome is based on a complex array of possible features and/or a pathogenic variant in the PTEN gene [4]. The sensitivity and specificity of the intramucosal lipoma in Cowden syndrome were recently described by Caliskan et al., wherein 32% of intramucosal lipomas occurred in patients either with definitive Cowden syndrome or with multiple Cowden criteria, while the remaining 68% were attributed to sporadic findings [1]. To our knowledge, intramucosal lipomas have not been reported in association with any other disease, cancer, or syndrome.
Li-Fraumeni syndrome is a highly-penetrant, autosomal dominant syndrome that predisposes affected families to multiple cancers, frequently at an early age. The syndrome is associated with pathogenic germline TP53 mutations. These families experience an estimated 73% lifetime risk of cancer in males and 100% in females [9-11]. The classic disease profile of Li-Fraumeni syndrome consists of “core” tumors, including sarcomas, leukemia, brain tumors, breast carcinoma, and adrenal cortical carcinoma. These core cancers make up 77% of all cancers in Li-Fraumeni syndrome families; the remaining 23% non-core cancers are highly variable [12]. Non-core Li-Fraumeni syndrome tumors have been found most frequently in the lung, stomach, ovary, colon, skin, and endometrium [14]. Gliomas are the most common form of brain tumor found in Li-Fraumeni syndrome [13] and were present in three 1st degree family members in this kindred.
Cancers in affected Li-Fraumeni individuals frequently manifest at young ages, with a reported 30% of cancers developing before age 20 and 36% developing in the third and fourth decades [14]. Many patients with Li-Fraumeni syndrome develop multiple cancers over their lifetime [15]. Due to the high incidence of neoplasia in these patients, frequent and thorough preventative cancer screening is necessary [16].
Multiple guidelines have been created to identify families with possible Li-Fraumeni syndrome. Most recently, the revised 2009 Chompret criteria provide guidance for TP53 mutation testing, with an estimated sensitivity of 35% [12]. These criteria recommend mutation testing for patients who 1) develop a Li-Fraumeni syndrome core tumor before age 46 and have a 1st or 2nd degree relative with an early onset LiFraumeni syndrome core tumor, 2) develop multiple tumors, two of which are a Li-Fraumeni syndrome core tumors and one of which is early onset, or 3) develop an adrenocortical carcinoma or choroid plexus brain tumor [12].
Although colonic neoplasia is not included as one of the “core” tumors in Li-Fraumeni syndrome, a variety of colonic pathologies are often present in these patients. Rengifo-Cam et al. found that 48% of Li-Fraumeni syndrome patients undergoing colonoscopy had colonic lesions, including tubular adenomas, hyperplastic polyps, high-grade dysplasia, and sessile serrated lesions [17]. Intramucosal lipomas were not reported in this series or elsewhere to our knowledge. An estimated 4% of Li-Fraumeni syndrome patients develop colorectal cancer before age 50 [18]. Another study found that 8.6% of Li-Fraumeni syndrome patients were diagnosed with either colorectal cancer or an adenomatous polyp with high-grade dysplasia before age 50, and that half of the diagnoses occurred before age 35 [19]. Due to this increased risk for early-onset colorectal cancer, colonoscopic screening every 2-5 years beginning at age 25 has been recommended for Li-Fraumeni syndrome [16,20].
This work was approved by the University of Utah Internal Review Board and includes informed patient consent. This 38-year-old man presented with a left temporal brain anaplastic astrocytoma, underwent complete resection, and made a successful recovery with adjuvant chemotherapy. He is now without disease at 29 months of follow-up. A strong family history of cancer, including both a sister and mother with glioblastoma multiforme, as well as ductal carcinoma in situ of the breast in the mother, prompted genetic investigation for a possible hereditary cancer syndrome. Next-generation sequencing of the following genes was performed by Ambry Genetics (Aliso Viejo, CA): AIP, ALK, APC, ATM, BAP1, BARD1, BLM, BRCA1, BRCA2, BRIP1, BMPR1A, CDH1, CDK4, CDKN1B, CDKN2A, CHEK2, DICER1, EPCAM (deletion/duplication only), FANCC, FH, FLCN, GALNT12, GREM1 (deletion/duplication only), HOXB13, MAX, MEN1, MET, MITF, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1, NF2, PALB2, PHOX2B, PMS2, POLD1, POLE, POT1, PRKAR1A, PTEN, PTCH1, RAD50, RAD51C, RAD51D, RB1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, SMARCB1, SMARCE1, STK11, SUFU, TMEM127, TP53, TSC1, TSC2, VHL, XRCC2. This testing revealed a likely pathogenic p.H193Q (c.579T>A) variant in TP53. No pathogenic/likely pathogenic variants were detected in PTEN. Subsequent testing of multiple affected family members discovered
the same pathogenic TP53 variant in the patient’s mother, two brothers and three children, confirming the diagnosis of Li-Fraumeni syndrome (Figure 1).
As part of Li-Fraumeni cancer screening, the patient underwent upper endoscopy and colonoscopy. A single colonic polyp from the sigmoid colon was biopsied and identified as an intramucosal lipoma. The lipoma diagnosis was confirmed by characteristic eggshell S100 immunostaining of adipocytes. The only other TP53 variant carrier in the family to have colonoscopy was the proband’s mother. Colonoscopy and pathology reports identified a tubular adenoma and hyperplastic polyp, but no intramucosal lipoma.
We report the first case of a colonic intramucosal lipoma in a Li-Fraumeni syndrome patient. Two possibilities exist for this rare finding. The polyp could either represent a new component of the colonic phenotype of Li-Fraumeni syndrome, or it may more simply represent an unrelated and sporadic intramucosal lipoma, which have been described. In a series of 25 patients with intramucosal lipomas, 68% were considered to be sporadic, without genotypic or phenotypic evidence of Cowden syndrome [1]. The coincidence of these two rare findings in our proband, namely Li-Fraumeni syndrome and an intramucosal lipoma, is however notable
This is the first report of this polyp in a Li-Fraumeni syndrome patient. Intramucosal lipomas are difficult lesions to diagnose due in part to their rarity, but also due to their very common histologic mimic, an artifact of colonoscopy known as pseudolipomatosis. Intramucosal lipomas, therefore, are often overlooked, but can be confirmed by a highly specific S100 immunohistochemical “eggshell” staining pattern. When found, intramucosal lipomas raise suspicion for Cowden syndrome, which is present in an estimated third of patients [1]. However, the occurrence in a family with Li-Fraumeni syndrome highlights the importance of routine collection of family history on all patients, in an attempt to identify previously unknown associations and phenotypes, and for referring for testing patients with a personal or family history suggestive of inherited cancer predisposition.

![]()

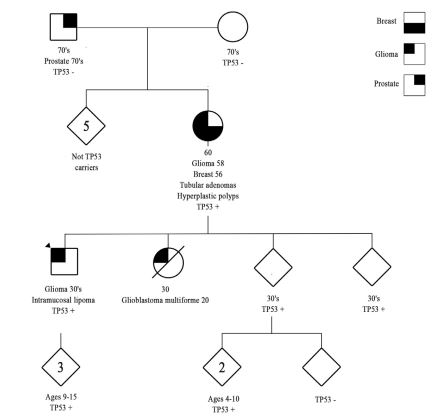 |
| Figure 1: Genetic pedigree of this Li-Fraumeni syndrome family, revealing striking glioma brain tumor penetrance in three 1st degree relatives and the first reported intramucosal lipoma of the colon in the proband (arrowhead). TP53 - = absence of TP53 germline mutation; TP53 + = presence of pathogenic TP53 germline mutation |
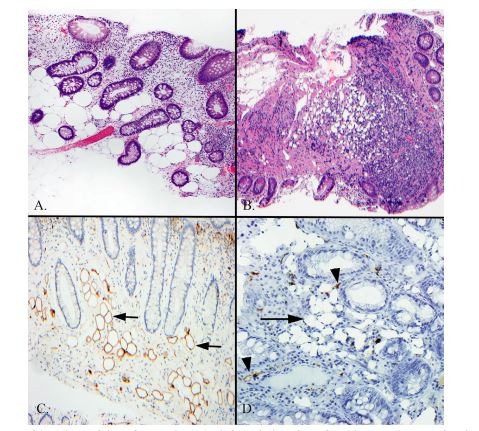 |
| Figure 2: (A) Intramucosal lipoma of the colon with large fat vacuoles (open holes) of relatively uniform shapes and sizes within the lamina propria, surrounding and between colonic mucosal crypts (hematoxylin and eosin stain); (B) Pseudolipomatosis coli from the commonplace colonoscopic insufflation artifact, mimicking an intramucosal lipoma, but showing in contrast, tiny and more irregularly sized collections of air bubbles (open holes) in association with a mucosal associated lymphoid aggregate (hematoxylin and eosin stain); (C) S-100 immunohistochemical staining of a true intramucosal lipoma revealing the characteristic and delicate brown egg-shell or rim staining pattern surrounding the full perimeter of fat vacuoles or holes (S100 immunohistochemical stain with diaminobenzidine brown chromagen and hematoxylin counterstain); (D) S-100 immunohistochemical staining of pseudolipomatosis coli showing in contrast, the lack of brown rim staining on the air bubbles (arrow) that mimic true fat. Note also at the two arrowheads showing brown S-100 positive lamina propria dendritic cells, which are virtually always present in normal colonic mucosa. Dendritic cells are identified by their irregular short, pointed and delicate cytoplasmic processes that are morphologically quite distinct from the circumferential eggshell pattern of adipocytes, but that could be misconstrued as true positivity in fat when scattered among the air bubbles of pseudolipomatosis (S100 immunohistochemical stain with diaminobenzidine brown chromagen and hematoxylin counterstain) |




































