Open Access
Research Article
Max Screen
ISSN: 2638-082X
Copyright: © 2021 Yalcin EB. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The ability to measure structural and functional alterations in cellular and tissue lipids with small footprint, accessible instrumentation has sparked interest in their role in disease pathology. However, various lipidomic analytical tools tend to be cumbersome and time-consuming. A rapid, accurate, and straight forward peak alignment software routine would greatly facilitate the analysis of large datasets, such as those produced by matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). Herein, we describe a novel Rapid Peak Alignment Method (RPAM) which allows untargeted analysis of lipids expressed in brain white matter following chronic ethanol exposure in an established experimental model. The RPAM outputs data comparable to manual peak alignments but the processing time requires only 90 minutes instead of 8-10 hours. This method is readily adapted to a broad range of models, tissue types, and human diseases.
Keywords:RPAM; MALDI-TOF; Untargeted Lipid Analysis; Peak Alignment
List of abbreviations:MALDI-TOF MS: Matrix Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry; m/z: Mass-To-Charge Ratio; RPAM: Rapid Peak Alignment Method
Matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) has become a standard tool for proteomics, lipidomics, metabolomics, and biochemical histopathology [1-3]. High sample throughput and speed make MALDI nearly indispensable for comparative untargeted lipidomics studies. This approach has been broadly used to identify the altered lipid patterns in neurodegenerative diseases such as Alzheimer’s disease, vasculature dementia, and stroke to better understand disease pathogenesis [4]. However, manual alignment of the vast data arrays that include hundreds to thousands of individual lipid species [5,6] is extremely time-consuming.
Although software such as ClinProTools v 4.0 (Bruker Daltonics, Billerica, MA) exists for handling large datasets [7,8], the results are not comprehensive because low-intensity peaks and peaks not expressed in all samples are excluded from the alignments. Such data compression may obscure critical disease-linked distinctions. Other approaches such as manual peak alignment or programs written in the R software environment produce complete sets of aligned peaks (mass- to-charge ratio (m/z) values) together with their corresponding intensities at the expense of a researcher's time [6,9,10] (see Supplementary Information). Herein, we describe a novel method of peak alignment termed “Rapid Peak Alignment Method” (RPAM) that was built on Microsoft Excel 365 (Microsoft Corporation, Redmond, WA, USA) platform for immediate portability to most laboratories. RPAM can be used for the comparative untargeted lipidomics analysis of multiple datasets and yields high-quality results in a fraction of the time required for manual data processing and without the data loss that occurs with compressive algorithms.
The Rapid Peak Alignment Method (RPAM) consists of three stages. The first step involves the alignment of a high throughput dataset in an automated fashion. The mass-to-charge (m/z) values of the peaks are rounded to the nearest integer values (e.g., 834.4 becomes 834), and the same integer values from different samples are aligned within the same row in excel. The second stage includes manual fine-tuning of errors in the alignment that might occur when there are two peaks with the same integer but different decimal points (e.g., 834.1 and 834.4) within the same sample. The third stage allows revision of the aligned data based on the standard deviation of the m/z values higher than 0.25.
Stage 1: Automated High Throughput Peak Alignment(Figure 1)
1. Copy and paste the mass-to-charge (m/z) values of the first sample into the “Data Input” sheet under the table titled “Sample 1”
(first cell: B3, column titled “m/z”). Paste the corresponding average peak intensity values into the same table (first cell: D3, column
titled “intensity”).
2. Repeat step 1 for subsequent samples. Our method can accommodate up to 40 samples.
3. Navigate to the “Copy This” sheet. The program automatically transfers the data set to this sheet starting with m/z values detected at
300 Da and increasing by 1 Da for each subsequent row. Note that the data can be located far down the sheet (i.e., m/z values of 600-
1200 Da mass range will appear between rows 300 and 700). Copy the entire data set starting with cell A2 (column titled “Sample 1”)
and including all m/z and intensity values for each sample.
4. Navigate to the “Paste Here” sheet. Use the “paste value” command to paste the copied data into the sheet on cell D2 marked with an “x”.
5. Select the entire data set together with the mass deviation, average m/z, and average intensity columns starting with cell A3.
6. Navigate to the insert tab and select table. Check “My table has headers” when prompted and click “Ok”.
7. Select the first three cells (A4, B4, and C4) and distribute the functions under each header to the bottom of the table.
8. Once the functions are distributed, click on the “average m/z” (located on cell B3) to sort the table from smallest to largest number.
Stage 2: Manual Fine Tuning (Figure 2)
1. Navigate to the “Error Identification” sheet. In this sheet, the first column (column A) represents the rounded average m/z values
and the subsequent columns represent the number of the specific m/z values within the samples. For example, if a sample contains
two m/z values that are rounded to the same integer (e.g., 834.1 and 834.4), the cell corresponding to that particular m/z will
be marked as 2 and highlighted in red. Note that the “Error Identification” sheet is a table, and each sample column can be sorted
from largest to smallest to help with identifying errors.
2. If a sample column has a red cell, navigate to the “Data Input” sheet. Identify the exact m/z values that have the same rounded
integer number and different decimal numbers.
3. Once all missing m/z values are identified, navigate to the “Paste Here” sheet and insert rows where the values belong.
4. Return to the “Data Input” sheet. Copy the first missing m/z value. Navigate to the “Paste Here” sheet and use the “paste value”
command to paste the m/z value into the proper row. Repeat the same task for the corresponding intensity value. Copying and
pasting the m/z and intensity values must be done independently.
5. Repeat step 4 for all missing m/z values.
Stage 3: Revision(Figure 2)
1. Once all data have been entered in the “Paste Here” sheet, examine the mass deviation function (column A) to help identify rows
where peaks could be misaligned. Any row with an average mass deviation higher than 0.25 will be highlighted in red. (Note: The
mass deviation value can be adjusted in the conditional formatting menu).
2. If a row has the standard deviation value highlighted in red, check to make sure the m/z values are properly aligned. If an m/z
value does not belong in the row, check the neighboring rows (±1 Da) and move the m/z value and corresponding intensity to
the correct row.
3. If the m/z value cannot be aligned to a neighboring row, insert an additional row above or below, and add the m/z value and
corresponding intensity to that row. Remember to redistribute the functions in the first three columns after making changes in the
alignment.
To examine the accuracy and speed of our Rapid Peak Alignment Method (RPAM), we aligned an existing data set generated in our laboratory using 1) RPAM, 2) ClinProTools software version 4.0 (Bruker Daltonics, Billerica, MA), 3) manual alignment, and 4) R software peak alignment algorithm. The data set includes a total of 24 rat frontal lobe samples imaged in the negative ion mode between 600 and 1200 Da mass range with an Ultraflextreme MALDI-TOF mass spectrometer (Bruker Daltonics, Billerica, MA).
The results depicted in Table 1 demonstrates that the manual alignment method required 8-10 hours to complete the peak alignment followed by RPAM, which required 90 minutes, while ClinProTools and R software were able to complete alignment within seconds. In the aligned data set, the total number of peaks in all samples were detected as 2595 in manual and RPAM, whereas this number reduced to 2400 and 634 in ClinProTools and R, respectively. For tentative lipid assignment, the m/z values of ions were searched against previously identified ions in our laboratory or other published reports. The number of ions that were assigned to a specific lipid species were higher in manual (244) and RPAM (246) relative to R (165) and ClinProTools (63). The average mass deviation of the aligned peaks was 0.07 in manual method, 0.03 in RPAM, and 0.05 in ClinProTools. No mass deviation was observed in R peak alignment.
We introduce a Rapid Peak Alignment Method (RPAM) built on Microsoft Excel 365 to facilitate analysis of high throughput MALDI-TOF data. Inter- and intra-group differences in peak profile compositions require peak alignments for untargeted lipidomics analysis [6,10,11], yet there is a limited body of literature for the peak alignment of MALDI-TOF data. Previous studies utilized ClinProTools (Bruker Daltonics, Billerica, MA) [7,8] or other methods such as manual alignment and programs written using the R software environment [6,9-11].
The primary drawbacks of existing peak alignment methods include the lengthy time and challenges to achieving accuracy and comprehensive rather than compressed results. The current software failed to provide a complete set of aligned peaks. In ClinProTools, data pre-processing excludes low intensity ions resulting in fewer peaks in the aligned data set. In R, the lower number of peaks is related to alignment of only a few samples, not the entire data set as the program crashes due to lack of random-access memory (RAM). Furthermore, these incomplete data sets presented inconsistencies between the number of lipids tentatively assigned resulting in fewer lipids in ClinProTools and R relative to RPAM. The discrepancy between the number of lipids identified in manual and RPAM was due to misaligned peaks in the manual alignment. These problems are resolved with RPAM. Although RPAM takes longer than the instantaneous alignment methods provided by ClinProTools [7] or R (see Supplementary Information), the final results are more granular (detailed) rather than compressed. Data compression corresponding to missing or low-profile peaks may be critical to understanding the effects of disease states. Future directions will focus on fully automating the RPAM algorithm, possibly through the use of machine learning.
This paper demonstrates a new peak alignment strategy to analyze high throughput data sets acquired with MALDI-TOF systems. The comparison of four alignment methods revealed that RPAM can align high throughput MALDI data sets within 90 minutes, with a higher accuracy than manual alignment and without the pitfalls of data compression associated with many commercial software programs.
https://annexpublishers.com/articles/JBMT/Supplementary-Information.pdf
Authors’ Contributions:WP developed the RPAM; WP and EBY evaluated RPAM accuracy and conducted comparative analysis with other methods; WP, EBY, and SMdlM wrote and revised the manuscript.
Declaration of Conflicting Interests:The authors declare no conflicts of interest.
Funding:Supported by the VA Biomedical Laboratory Research and Development (BLRD) Career Development Award-2 1IK2BX004961 (EBY), R01- AA11431 (SMdlM), and R01- AA028408 (SMdlM).

![]()

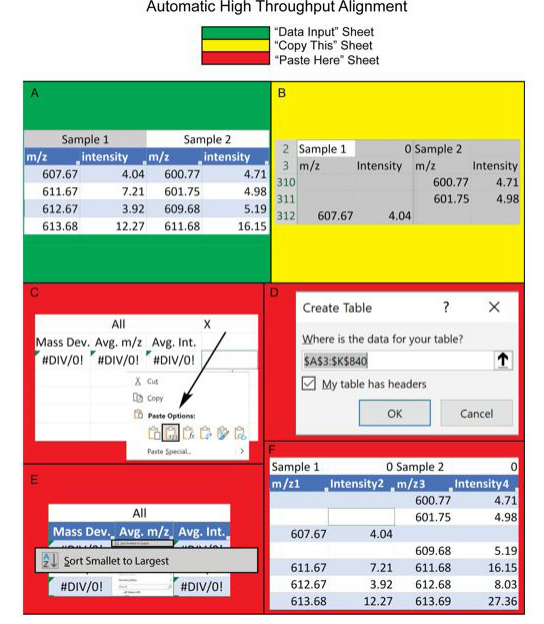 |
| Figure 1: : Rapid Peak Alignment Method (RPAM) Diagram- Stage 1: Automated High Throughput Alignment Workflow. (A) Raw data are copied into the "Data Input" sheet, with the mass-to-charge ratio (m/z) and intensity values entered in separate columns; (B) "Copy This" contains the preliminary alignment. In the example data shown here, the first peak (m/z 607.67) of sample 1 is located in row 312. Data from this sheet is copied, including the sample headers (Sample 1, Sample 2 etc.); (C) The data are pasted onto the "x" mark in the "Paste Here" sheet using the "paste value" command; (D) The entire data set is highlighted including the sample headers and the first three columns to create a table with headers in the "Paste Here" sheet; (E) Data within the table are sorted by average m/z in ascending order; (F) The preliminary alignments are generated such that peaks with the same m/z values are aligned within the same row |
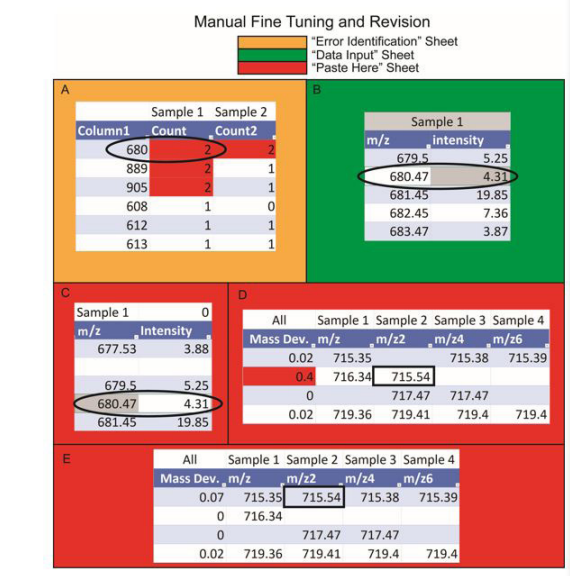 |
| Figure 2: Rapid Peak Alignment Method (RPAM) Diagram- Stages 2-3: Manual Fine Tuning and Revision Workflows. (A) The "Error Identification" sheet reveals m/z values that are missing from the preliminary alignment due to rounding of m/z values to the same integer, e. g. 679.5 and 680.47 are both rounded to 680; (B) The "Data Input" sheet contains the two different m/z values (679.5 and 680.47) detected in the same sample (Sample 1) but rounded to the same integer (680); (C) The "Paste Here" sheet contains the smaller m/z value (m/z 679.5) after automated alignment, whereas the greater m/z value (680.47) must be inserted manually; (D) Following the addition of missing data, the alignment is revised by checking the standard deviation of m/z values. If mass deviation is greater than 0.25, the cell is highlighted in red. The misaligned m/z values, e.g. 715.4, are transferred to the correct row with matching m/z values; (E) The final peak alignment demonstrates the correct placement of m/z values. Note that the peak intensity values are hidden in the figure for simplicity |
|
Manual |
RPAM |
ClinProTools |
R |
Time Required for Analysis |
8-10 Hours |
90 Minutes |
<1 Minute |
< Minute |
# Peaks Detected |
2595 |
2595 |
2400 |
634 |
# Lipids Detected |
244 |
246 |
63 |
165 |
Standard Deviation of m/z Values |
0.07 |
0.03 |
0.05 |
0 |




































