Open Access
Research Article
Max Screen
ISSN: 2455-765X
Copyright: © 2018 Shang N. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Attribution of etiology for disease syndromes is critical to guide appropriate public health interventions. Partial latent class analysis model (pLCM) methods have recently been developed to address this area of research; however, model parameters, assumptions, and performance are not well understood for the general etiology problem. Here, we first establish a relationship between the etiology proportions defined by pLCM and those defined by population attributable risks (PAR) under a unified probability structure. We performed simulation studies to characterize scenarios where pLCM models may not yield reliable estimates and to illustrate the strengths and limitations of pLCM. We identified mitigation strategies for scenarios in which model performance was not optimal. Based on these results and the theoretical examinations of pLCM parameters and assumptions from the population attributable risk point of view, we propose systematic strategies to enhance etiology research, drawing upon the recent experience of the Aetiology of Neonatal Infections in South Asia (ANISA) study.
Keywords: Disease Etiology; Population Attributable Risks; Latent Class Analysis; Gibbs Sampler
Determining etiology of infections is complex as the laboratory test based evidence of pathogenic infection is often nondeterministic. Recent advancements in molecular testing techniques provide accurate laboratory evidence for pathogen presence in clinical specimens. However, presence of a pathogen does not necessarily imply that the pathogen causes the disease, especially for pathogens that may have a carriage state in the healthy population. Standard epidemiologic comparisons of laboratory detection of pathogens among samples from disease cases and healthy subjects become complex to interpret when pathogen carriage rates are high and laboratory tests are imperfect.
In recent years, studies have used an attributable etiologic fraction (PAR) approach that applies the concept of population attributable risk to the etiology question [1]. These methods implicitly assume perfect sensitivity of all laboratory tests; i.e., true exposure to the pathogens is 100% detectable by the tests. Furthermore, the extension of the population attributable risk concept to the estimation of the attributable fraction of more than 1 pathogen simultaneously through logistic regression lacks statistical or epidemiological justification and often results in confusing conclusions [1-3]. It is also not possible to combine results for the same pathogen across multiple specimens, or to provide an etiologic attribution at the individual level for individual cases. To address some of these limitations, Wu et al. developed a partially-Latent Class Model (pLCM) for the Pneumonia Etiology Research for Child Health study to attribute etiology and characterize the associated pathogen proportions among pneumonia cases in children under 5 years of age [4]. Implementation of pLCM to the PERCH (Pneumonia Etiology Research for Child Health) project was presented in Knoll et al. [5].
However, epidemiological interpretations of pLCM parameters and model assumptions raise questions. For example, under what conditions that the etiology proportions defined by pLCM relate to that defined by PAR? What are the practical implications of violating these conditions? Resolution of these issues would aid identification of pathogens that could be appropriately studied by pLCM. Further, the numerical performance of pLCM has not been extensively studied yet. The successful application of pLCM to the PERCH study may have been fostered by key study features not always achievable in etiology studies: case definitions specific for pathogenic infection; an adequate sample of health subjects providing accurate estimation of false positive rates; an adequate sample of cases with positive gold standard results; and prior knowledge of the sensitivity of the employed laboratory tests) [5].
In section 2 of the paper, we present a unified probability structure for pathogenic infections that assumes exposure to the pathogens can result in a disease-free carriage status. This state can be identified by one or more laboratory tests, and such carriage increases the likelihood of developing disease. Under this structure, we show that the pLCM defined etiology proportions are approximately equal to that defined by PAR under a few assumptions with clear epidemiological interpretations.
Then in section 3, we conduct and summarize a series of simulation experiments that shed light on when and how pLCM produces accurate estimates, which allow for further understand of the pLCM model. Together with our epidemiologic interpretations of the model parameters and assumptions, these simulation experiments provided general insights and guidelines on design and analysis of etiology studies using the pLCM method. We summarized those insights and guidelines in appendix with a description of our implementation practice strategies in a large community-based study, Aetiology of Neonatal Infections in South Asia (ANISA). The real data application of our methods was presented and published in the Journal of the Lancet [6].
Two approaches to define pathogen etiology proportions for one single pathogen: A disease etiology study could be a case only study if there was a gold standard to identify each case’s true pathogenic causes. In reality, such a gold standard is usually not available. For example, currently available laboratory tests are only capable of detecting presence of pathogens in the collected specimen, and/or may fail to detect all possible infections. Standard epidemiological strategies address the issue through collecting samples from healthy subjects and then comparing laboratory detection of pathogens among samples from disease cases and healthy subjects. Two different approaches were developed based on different interpretations of the strategy.
Given a disease, a pathogen and a corresponding laboratory diagnostic test that was well calibrated with 100% laboratory specificity, the population attributable risk (PAR) approach defines the relationship of the pathogen to the disease through considering exposure to the pathogen as a risk factor of developing disease. This approach uses the laboratory test result as indication of the exposure. The pathogen proportion is then defined as the proportion of disease cases being prevented if the pathogen exposure was removed completely form the study population. For example, if the disease rates are le and lu in the exposed and unexposed respectively, then the pathogen’s proportion of the disease is defined as:
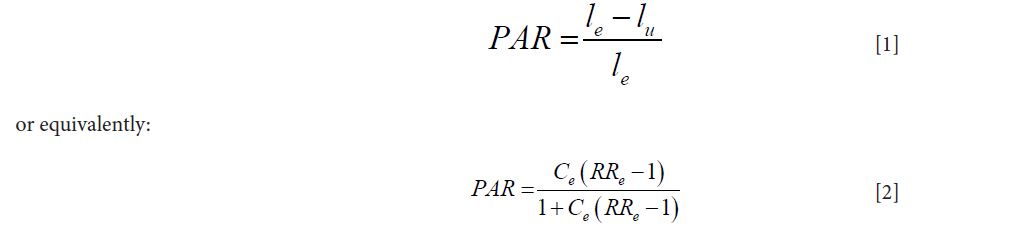
where Ce is the proportion of subjects with exposure to the pathogen, or with positive laboratory test results, in the study population RRe expresses the risk of developing disease among those with exposure to the pathogen, relative to those without such exposure. Since, in general, the laboratory test is not 100% sensitive, calculated from the positive laboratory test results would underestimate the true exposure proportion (assuming perfect specificity). The corresponding PAR estimation would then usually underestimate its true value.
Although PAR is a population based concept, in practice it has been applied to case-control study settings too, replacing the relative risk with the relative odds (or odds ratio), and replacing Ce with proportion of controls with positive test results. However, such approximation might cause some severe bias, especially when the laboratory test cannot detect non-disease status of pathogen carriage, and/or when the control samples are substantially different from the general study population due to matching of controls to the cases.
Another approach to define pathogen proportion is to extend the original laboratory test for pathogen appearance into a pseudo test for disease etiology and integrate it in a mixture model for cases. Here, all cases are divided into two groups: those with the pathogen as etiology cause and those with other causes. The etiology proportion is then the proportion of the group with the pathogen as etiology. However, since we can only observed the laboratory test results, not the group membership of the subjects, a pseudo binary test is constructed based on the laboratory test with true positive rate θ and false positive rate δ defined as
Note that this pseudo test is different from the original laboratory test. For example, the laboratory false positive rate (1 – specificity) is usually very small for our well-calibrated laboratory tests, but the false positive rate for the pseudo test could be very high as it measures the carriage status of the pathogen in study population. Also note that while this definition is based on cases only, sample from healthy subjects can be used to construct estimations for the usually unknown false positive rates.
Under this mixture model structure, if both θ and δ are known, then the pathogen proportion can be calculated with following formula:
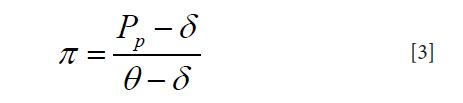
Where PP is the proportion of having positive laboratory test results among cases. Following lemma establishes a relationship between the etiology proportions defined by the two different approaches.
Lemma 1: If the false positive rate equals to the product of the true positive rate and the study population’s exposure rate to the pathogen, i.e.

then the population attributable risk as defined in equation [2] equals to the mixture component proportion defined in equation [3].
To understand what might be implied in equation[4] let’s consider a simple, yet common infectious disease development process in which exposure to pathogen results in a carriage state prior to developing disease. Now suppose the pathogen will appear in the collected specimen for subjects in either carriage or disease status, then the true positive rate is just the laboratory test’s sensitivity. Hence the right side of equation is the probability of being tested positive for subjects in the study population. The quantity only approaches the false positive rates (i.e., the left side of Equation) when the proportion of all carriers that would develop disease is low. In other words, if removing diseased cases from study population will not substantially modify carriage proportion Ce, then the two definitions of pathogen etiology proportions are approximately the same.
However, for some other pathogens there is no such a carriage status and/or the laboratory test cannot detect the status. The corresponding false positive rates are then equal to zero. Hence, Equation [4] cannot not be true. Consequently, the pathogen proportion defined via the PAR approach will not equal that defined by the mixture model. Pathogens with such characteristics will further challenge our interpretations of pathogen proportions when the two definitions are extended to a situation of multiple pathogens.
We now extend our definitions of pathogen proportions to multiple pathogen situation. Suppose there are K≥1 pathogens that may cause the disease. For pathogen k,k=1,...,K let Tk be an indicator variable for the result of a binary diagnostic test. As before, a positive test result (i.e. Tk = 1) indicates that the pathogen appears in the collected specimen (i.e. the subject is exposed to the pathogen).
In the literature, there are two ways to extend the PAR concept to multiple pathogens. For a subset of pathogens S, the first approach defines the combined etiology of the S pathogens as the proportion of cases being prevented should all pathogens in S being removed from the study population, with formula similar to Equation [1]. This approach neither offers an alternative formula such as the one in Equation [2] that relates PAR with relative risk, nor can it establish a relationship between the combined PAR with individual PARs. For example, it usually does not suggest additivity of the individual PARs:
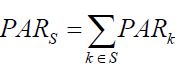
The other approach to extend PAR to multiple pathogens is to consider the K test results as K risk factors and apply logistic regression to obtain odds ratios for each factor. The model adjusted odds ratios are then utilized in Equation 2 (as approximation to the relative risk) to calculate individual “adjusted” etiology fractions. Unfortunately, such ad hoc approach does not retain good epidemiology interpretation of PAR. Furthermore, a few artificial adjustments are required. For example, to avoid possible negative values of “adjusted” etiology fractions, pathogens with odds ratios less than 1.0 are removed from the regression equation Additionally, the approach does not constrain the individual etiology fractions to be additive to the combined PAR and does not even constrain the overall sum of individual adjusted PAR to within 100%.
On the other hand, extension of mixture model approach to multiple pathogens is straightforward. All cases are divided into K + 1 groups corresponding to the K pathogens and one extra class of “Other/None” for cases caused by none of the K pathogens. Let

Our question is: under what conditions do the two approaches define similar pathogen proportions? More precisely speaking, when does a relationship as in Lemma 1 hold for multiple pathogens? Since the mixture model based definition implies additivity of individual pathogen proportions, we also explore the conditions that when PAR based definition possess such additivity. We answer those questions based on a common probability structure as introduced below.
Let the baseline disease rate, or probability of developing diseases in a given time period without exposing to any of the K pathogens, be P0. For a subset S of the K pathogens, let SpΔ be the incremental probability of developing the disease for subjects carrying all of the S pathogens, with the limitation that P0+ΔpS≤1.0 Also let CS be the proportion of the population exposed to all pathogens in S. When S contains only one pathogen such as pathogen k, the incremental probability of developing disease and the carriage rate are simply denoted as Δpk and CK respectively. Then following lemma says the additivity of the incremental risks implying additivity of the PAR defined pathogen proportions.

Examples of violation of the additivity of incremental probability of developing disease include pathogens that are very infectious, such as exposing to the pathogen would almost certainly imply disease. They also include pathogens that do not have a carriage status or the carriage status cannot be detected by the corresponding laboratory tests. For such pathogens, there will be no positive test results from healthy population. These are also the situations that a case control study design might not properly estimate the pathogen proportion using the PAR approach as the odds ratio would become infinity and the individual PAR as defined in Equation 2 would be 100%. Therefore such pathogens’ pathogen proportions might be estimated and interpreted alone with an understanding that their individual pathogen proportions are not additive to other pathogens’ pathogen proportions.
Lemma 3 For any given subset of pathogens S, let ΔpS and CS be the incremental probability of developing diseases and the proportion of study population exposed to all pathogens in S, respectively, then with following conditions:
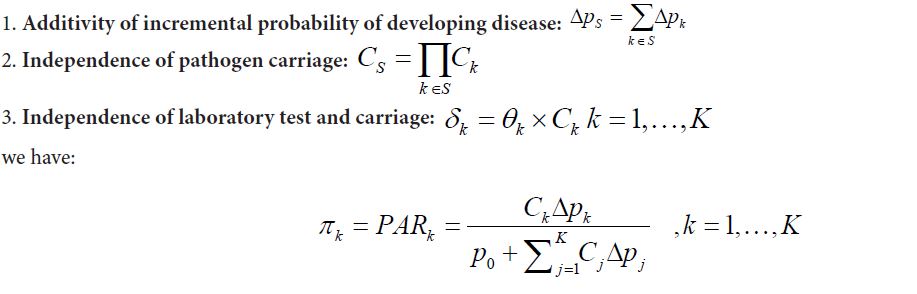
Similar to Lemma 1, condition 3 implies that for each pathogen, only a small proportion of exposed subjects will develop into disease, in other words kpΔ needs to be small for all pathogen k. In fact, if there are some pathogens with large Δpk, then it is likely that either condition 1 or condition 3 will become invalid. Therefore, in order to retain additivity of individual etiology proportions (required for mixture model approach) and to offer a proper epidemiology interpretations to the defined etiology proportion, we should be careful to include pathogens with large Δpk values in our model. Such pathogens may need to be studied separately with an understanding that their etiology proportions cannot be additive to other pathogens.
When the model parameters θK and δk, are unknown further assumptions are needed in order to estimate the etiology proportions πk in the mixture model . Let yik be the test results for case i and pathogen k using laboratory test Tk If we further assume conditional independence of the laboratory test results:

The parameters of the basic pLCM in Equation [7] can be estimated under a Bayesian analysis framework using conjugate priors for the parameters, for example, (K+1)-class Dirichlet distributions for the pathogen proportions and Beta distributions for the true and false positive rates, to construct a Gibbs sampler. Let Π=(π1,.....πk,πk+1),Θ=(θ1,....,θk and Δ=(δ1,....,δk) be the pathogen proportions, true positive rates and false positive rates respectively. For case i Let Yi=(Yi1,Yi2,....,Yik) be the observed binary results for the K tests and Zi=(Zi1,Zi2,....,Zik,Zi(k+1)) be the imputed pathogen classes from the

The parameters are updated by adding the number of cases assigned to each class to the corresponding prior distribution’s parameter of the same class.
Similarly, if we use a joint bivariate Beta distributions for the priors of TPRs and FPRs:

For example, the Beta parameters for the posterior distribution of TPRs will be updated by all cases assigned to the pathogen class: the number that tested positive will be added to the first parameter of the prior Beta distribution while the number that tested negative will be added to the second parameter.
The sampling distribution of the latent class Zi given observed data and the parameters Θ,Δ,Π, i.e.,f(Zi|Yi,Θ,Δ,Π) is proportional to a product of the pathogen proportions Π and a vector of weights: Wi=(Wi1,Wi2,....,Wik,Wi(k+1)) defined as:
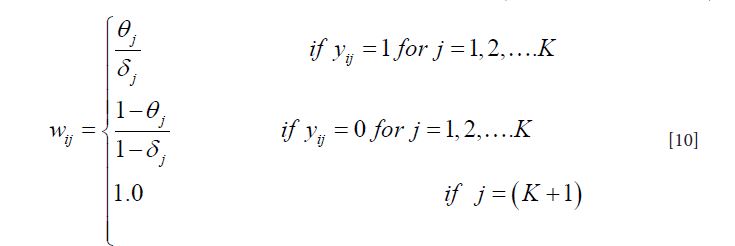
A random sample of Zi can then be drawn from the multinomial distribution proportional to Π*Wi.
The basic pLCM can be extended to situations where multiple pathogen-specific tests are performed for some of the pathogens. The only change is in Equation [10] where the weights will be multiplied for the performed tests, with an assumption that the test results are conditionally independent given the actual status of disease etiology
It is also straightforward to extend pLCM to situations with discrete strata of covariates, such as study site, age at enrollment, age group, and disease severity status. In such an extended model, the pathogen proportions and the false positive rates are allowed to change from strata to strata, reflecting variation of disease etiology and pathogen carriage rates. The true positive rates do not vary across different strata, since the case definition and laboratory procedures are identical, regardless of the covariate values.
While the primary objective is to estimate pathogen proportions at the population level, pLCM, as suggested by the implemented Gibbs sampler, addresses the question through iteratively identifying the etiology of individual cases, or equivalently imputing the latent classes at each iteration of the Gibbs sampler. Consequently, pLCM also establishes the individual etiology, in terms of probability that an individual case is due to each of the pathogen classes as presented by the posterior predictive distribution. Such individual pathogen probabilities, or the corresponding imputed latent classes, can be used to diagnose validity of pLCM model assumptions.
Simulation design and objective: The basic structure of pLCM including model equation, definitions of parameters, the Gibbs sampler implementation in estimating the model parameters, as well as extension of the basic model to more general situations are described above. Our primary objective was to understand the performance of pLCM in estimating pathogen proportion under a range of conditions. As we mentioned earlier, success of pLCM relies on using data of direct relevance to etiology (high quality control data so that false positive rates can be estimated accurately, and positive results from gold standard tests for true positive rates and pathogen proportions) to partially reveal the latent pathogen infection status of individual cases. Extra information, such as historical knowledge on accuracy of some laboratory tests of pathogens provides constraints that facilitate convergence of pLCM as demonstrated in the PERCH study [5].
Our simulation experiments were designed to explore the impact of such factors on pLCM accuracy and to describe conditions where pLCM performance may be unreliable.
The basic simulation design consisted of 9 pathogens with pathogen proportions increasing from 0 to 8% at 1% increments. The 9 pathogens account for 36% of the case population and the “Other/None” class accounts for the remaining 64%. The simulation generated 4,000 cases and 1,200 healthy individuals, and each simulation experiment was replicated 100 times. All simulations were run for 10,000 iterations with the first 5,000 as the burn-in period. We assessed model performance in estimation of individual pathogen proportions by a relative absolute deviance measure defined as
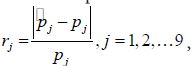 where pj is the true pathogen proportion and pj is the model estimate. We consider rj≥0.5 as poor estimation. If a large proportion of simulation replicates generate poor estimates, then we consider pLCM inappropriate for the specific simulation setting.
where pj is the true pathogen proportion and pj is the model estimate. We consider rj≥0.5 as poor estimation. If a large proportion of simulation replicates generate poor estimates, then we consider pLCM inappropriate for the specific simulation setting.
The first set of simulations assesses the impact of different combinations of true and false positive rates on pathogen proportion estimates based on the basic simulation design above, with one test per pathogen. The second set of simulations explores whether the introduction of multiple tests (with potentially different specimens) for a single pathogen would substantially improve the estimation. In this context, multiple tests refer to either different laboratory tests performed on the same specimen type (such as blood culture and molecular testing of nucleic acid extracted from blood), or the same assays run on different specimens from the same case (such as blood and nasopharyneal/oropharyngeal swabs); it does not refer to multiple replicates of the same assay. Since the regular latent class model can be applied to estimate a single pathogen proportion if there are at least three (conditionally independent) tests for that pathogen, we then compare the performance of pLCM with that of the regular latent class model when there are three separate tests for each of the pathogens [7]. Finally, we assess if one or more cornerstone pathogens will improve the overall estimation of pLCM by assigning pathogen status to a subset of cases with a high probability.
We used a Dirichlet(1,....,1) distribution-- the uniform distribution on the 10 dimensional simplex as the prior distribution for pathogen proportions, and used Beta(1/2,1/2)--the Jeffery non-informative prior for Bernoulli events—as the prior distribution for the TPRs [8]. This prior distribution setting is equivalent to adding a total of 10 pseudo cases, or one pseudo case per pathogen class, to the case population of 4,000, with 50% of chance to be tested positive for each of the laboratory tests. Overall, the contribution of the prior distribution to the pathogen proportions is very small and non-informative. The prior distributions for FPRs are constructed using the healthy individual data: the two parameters of the Beta distributions are simply the number of positive and negative test results among healthy individuals.
Performance of pLCM for different combinations of TPRs and FPRs: In the first experiment, we assessed pLCM performance assuming there was only one laboratory test for each of the 9 pathogens. We assume the 9 tests shared the same values of TPR and FPR. Three levels of FPR (30%, 10% and 1%) and four levels of TPR (40%, 60%, 85% and 95%) were examined for a total of 12 combinations. Based on the average pathogen proportions (over the 100 simulation replicates) and assessment of the relative absolute deviance, when the underlying noise (true FPR) was large, model estimates were poor (Table 1).
When the ratio of true TPR / true FPR was low, the estimated proportions were similar across pathogens regardless of the true underlying proportions (Table 1, first two rows). This suggests that noise (high FPR) masked the difference across pathogens even for some pathogens with moderate proportions, e.g. 8%. The smaller values of relative absolute deviance in Table 1 (such as at P8, TPR=0.4 and FPR=0.3) were artificial due to the fact that we have exactly 10 pathogen classes.
Model estimation of pathogen proportions improves as the FPR reduces; however, it is only when the true FPR=1% that the model estimates become acceptable with absolute bias less than 1.0% and the percentage of poor estimates less than 30%. Nevertheless, we observed that within the same FPR level, higher resolution of the laboratory tests or a higher value of the true pathogen proportion resulted in better pathogen proportion estimation. Finally, the model usually over-estimated pathogen proportions for pathogens with low true proportions. Bias was especially large for true pathogen proportions between 0 and 1%. Correspondingly, the proportion assigned to the Other/None class was usually under-estimated by pLCM.
Effect of multiple tests on single pathogen proportion estimation: For the same 9-pathogen design with 4,000 cases and 1,200 healthy individuals, we next considered three different tests named similarly to the actual tests used in ANISA, and with characteristics similar to some of the pathogen specific tests in ANISA [6]. The laboratory methods for molecular testing (TAC) and blood culture were described in Saha et al. [9]. The three tests are comprised of the blood TAC (test 1) (TPR=0.40, FPR=0.10); respiratory TAC (test 2) (TPR=0.85, FPR=0.10), and blood culture (test 3) (TPR=0.2, FPR=0.0). We then evaluated the accuracy of pathogen proportion estimates resulting from six models: blood TAC alone (model 1), respiratory TAC alone (model 2), blood TAC+blood culture (model 3), respiratory TAC+blood culture (model 4), respiratory TAC + blood TAC (model 5), and all three tests (model 6). The overall performance of the six models is presented in Figure 1. The percentage producing poor pathogen proportion estimates is summarized in Table 2.
Adding blood culture to a imperfect (TPR=0.85, FPR=0.10) test such as the respiratory TAC test greatly improved pathogen proportion estimates, while adding blood culture to a poor (TPR=0.4, FPR=0.10) test such as blood TAC did not result in accurate estimates. Moreover, combining the two TAC tests generated much better estimates than each test alone. The best performance was achieved when all three tests were used. Therefore, it is always beneficial for pathogens of key epidemiologic interest to incorporate multiple pathogen-specific tests into the study design, especially when a high FPR is expected.
For designs including three tests for a single pathogen, the pathogen proportion can be estimated by standard latent class analysis methods (such as the method in Latent Gold Statistical Package) [7]. However, information from other tests for other pathogens can provide additional improvement, although in some indirect way, regarding pathogen proportion estimations. For example, the probability of a case assigned to a given pathogen increases if the case tested negative for all other pathogens. Standard latent class analysis ignores such indirect information, while pLCM incorporates it naturally.
For the same simulation experiment above, we conducted separate latent class models for each of the 9 individual pathogens and compared the estimated pathogen proportions with those obtained from pLCM (Table 3). As anticipated, pLCM yielded more accurate estimates than standard latent class analysis.
The indirect effect of including multiple pathogens in pLCM: For a majority of viral pathogens and a few bacterial pathogens in ANISA only single tests were performed. Such pathogens cannot be studied individually using a traditional latent class modelling approach. While pLCM provides a general solution, Table 1 shows that if all tests were associated with high background noise, then the pathogen proportion estimates from pLCM might be questionable. However, if the pathogen list in the study contains some cornerstone pathogens with either multiple tests, small false positive rates or high true pathogen proportions, then attribution to the “cornerstone” pathogen class will be more accurate, improving estimation of true and false positive rates for the other pathogens lacking these desirable features.
In this simulation experiment, we created an input dataset with 8 pathogens (pathogen proportions 1 to 8%) and a single laboratory test each, and one with an additional 8 pathogens with the same pathogen proportions: 1 to 8%. These extra pathogens were each tested by all three tests introduced above (Table 2). We then estimated the pathogen proportions of the first 8 pathogens by pLCM including the 8 pathogens only and by pLCM including the original 8 plus the additional 8 pathogens. Either way, the input data for the first 8 pathogens was the same. The simulation results documented a clear improvement in accuracy of pathogen proportion estimates especially for pathogens with smaller true proportions (Table 4) (Figure 2).
Attribution of etiology for disease syndromes of importance is critical to guide appropriate public health interventions, but poses notable challenges from the laboratory, epidemiological and statistical perspectives. The problem is to deduce pathogen etiology from laboratory test results which only indicate presence of pathogens in the collected specimen, but do not serve as direct evidence of the cause of disease, particularly for pathogens with a carriage state.
While the PAR approach compared pathogen positive laboratory test results between cases and healthy individuals, pLCM compares true and false positive rates based on unobserved (latent) etiology status. The direct modeling of the latent etiology status (through a linear mixture model) has many advantages over the PAR approach, including allowing imperfect laboratory tests, direct modeling of multiple pathogens, incorporating multiple tests for a single pathogen, and more efficient use of historical knowledge through prior distributions. Hence pLCM provides a more versatile methodologic framework than previously available [1,8,9].
Despite its statistical appeal, pLCM does not offer clear epidemiological interpretations for its parameters and model assumptions, as the solutions from pLCM have not yet been mapped to some existing disease etiology concepts. This approach possesses two important assumptions: additivity of pathogen proportions from different pathogens and the conditional independence assumption. Both are difficult to examine (and thus verify) from an epidemiological point of view. As a result, applying epidemiological knowledge to reduce violations of pLCM assumptions remains challenging. In this paper, we provided a general probability structure for pathogen etiology problems. We showed that under certain assumptions, the pathogen proportions defined by the mixture model approach agrees with those defined by the population attributable risk approach approximately. Those assumptions include: exposure to a pathogen increases the probability of disease development and exposure to multiple pathogens results in an increase in probability of disease development that is additive over the individual pathogens. Pathogens that violate those assumptions were not suitable for pLCM model and we should study them and interpret their etiology proportions separately.
In additional to lacking epidemiology interpretations, the structure of basic pLCM requires estimation of many model parameters. The model parameters can easily become non-identifiable due to a large number of parameters and relatively few degrees of freedom (as the number of distinctive combinations of laboratory test results) [9]. Existing standard pLCM methodology suggests dealing with the identifiability issue through inclusion of a large and representative healthy individual sample, and a few cases with gold standard pathogen test results [4]. However, as our simulation experiments suggested, these conditions alone do not guarantee correct estimation of pathogen proportions. Successful application of pLCM requires more information garnered either from study design or historical knowledge.
One way to include historical knowledge on laboratory test accuracy is to use an informative prior distribution for the true positive rates [5]. However, when such information is unavailable or unreliable, inclusion of a few cornerstone pathogens is just as important in improving the performance of pLCM. Because attribution to the cornerstone pathogen classes is highly accurate, this improves the accuracy of estimates of other pathogen proportions. For example, if a case tested positive for a cornerstone pathogen, then the case can serve as a negative control for other pathogens, hence improving the accuracy of the estimation of false positive rates. Our simulation experiments clearly demonstrate such indirect benefits from pLCM.
It is also critical to exclude pathogens likely to result in non-identifiability. Most commonly, this includes pathogens with a single laboratory test that yielded very few positive results, as well as pathogens with high carriage rates in the study population or with a low ratio of true to false positive rates. Pathogens with these attributes should be carefully examined and excluded from pLCM.
pLCM is a powerful advance in etiology attribution, and estimates pathogen proportion accurately under a wide range of conditions; however, there are important analytic pitfalls to be aware of. Many of these can be mitigated through the approaches described in this paper. Nevertheless, there is no systematic approach to examine all identifiability issues. The procedures we suggested in this paper should be regarded as preemptive strategies to prevent model identifiability issues. Future research is needed for developing tools that examine these issues in a more systematic way [10-12].
The authors thank Dr. Maria Knoll, Dr. Zhenke Wu, Dr. Scott Zeger, and the entire Pneumonia Etiology Research for Child Health (PERCH) team for their invaluable advice and assistance with the development of the ANISA statistical methodology. We thank Dr. Samir Saha for his guidance and leadership of the ANISA study, as well as the entire ANISA team. Wementor thank Dr. Cynthia Whitney for her leadership and mentorship of the US Centers for Disease Control and Prevention (CDC) members of the ANISA team.
For a single pathogen k, let CK be the carriage rate in the study population and Δpk be the incremental probability of developing disease from carrying the pathogen. Also, let P0 be the background disease developing probability without carrying the pathogen. Then the expected risk of developing disease in the population will be le=p0+CkΔpkand lu=p0. Therefore, the PAR as defined in Equation 1 will be:
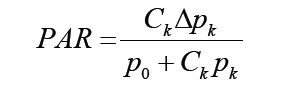
However, among the cases, the proportion of carrying the pathogens and hence will be tested as positive by TK will be:
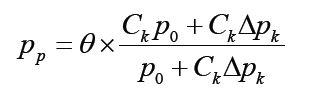
Where θ is the true positive rate of the laboratory test. Substituting p_p and the relationship δ=θ×Ck into the definition in Equation 3, we have the etiology proportion defined by the mixture model approach as:
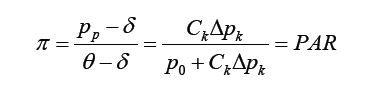
That conclude Lemma 1.
TO show Lemma2 without loss of generality, we consider two pathogens k1 and k2 with probability of carrying both,only k1,only k2and carrying neither as C11,C10,C01and C00respectively.The probability of developing diseases under the four scenario will be p0+Δp1+Δp2,p0+Δp1,p01+Δ2and p0 accordingly. From here we can calculate the PAR for K1 and K2 individually, and then the combined PAR for both pathogens, such as:
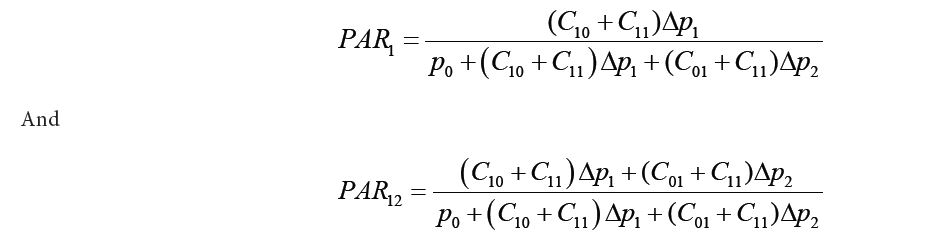
Hence,PAR12=PAR1+PAR2
Using the same two pathogens scenario, if we further have C11=C1×C2 and C10C1×(1-C2), etc., namely the carriage of the two pathogens were independent, then Lemma 3 follows the same deduction as above.
In addition to a large and representative healthy individual sample and a sufficient number of cases with a positive gold standard test results, which are not always feasible, our simulation results suggest that the performance of pLCM will be greatly improved if there are a few cornerstone pathogens in the pathogen list. Those pathogens have either multiple laboratory tests and/or the tests have any of the following desirable features: low false positive rates (as observed from the healthy individuals), a high ratio of true positive rate to false positive rate, and high true pathogen proportions. In some cases, pathogens with some characteristics should be excluded from pLCM modeling. For example, if a pathogen is tested by a single laboratory test and that test yielded very few positive test results, then the pathogen should be excluded, as the few positive cases from the test are not sufficient for accurate estimation of the true positive rate. More technically, the degrees of freedom of pLCM are based on the number of different combinations of the laboratory test results (across all pathogens) rather than the sample size of cases. If adding one pathogen into pLCM will only increase the number of combinations by at most two, then the addition of the pathogen should be rejected as each pathogen addition introduces at least two extra parameters (true positive rate and the pathogen proportion, assuming the false positive rate can be estimated from the healthy individuals) into the model. Based on these observations, in ANISA [12] we designed a strategy to exclude pathogens with single laboratory tests that yielded limited positive results. The strategy followed a stepwise selection procedure:
a: Established a list of cornerstone pathogens: For ANISA we had six that were tested by both respiratory and blood TAC. Four of those were also tested by blood culture. For other etiology studies, the list might be established by other desirable properties, or some subjective knowledge of pathogen proportions and laboratory tests; however, it is advisable at the study design stage to identify a few key pathogens of interest and ensure these will have multiple test results.
b: Added any pathogens that were tested by blood culture and blood TAC with positive blood culture result For example, in ANISA 6 additional pathogens were tested by blood TAC (a poor test) and blood culture (a gold standard test where positive results can be considered as confirmation of infection by the pathogen). If any of these pathogens had positive blood culture results, the pathogen will be included in pLCM. Any pathogens that were only tested by blood culture and yielded at least one positive result were not included in pLCM but were included in the overall etiologic attribution as described in Saha et al. [12].
c: Evaluated remaining pathogens through a stepwise selection procedure
1) At each step, we selected the pathogen from the candidate list that gave the largest increase in the number of distinct test result combinations.
2) We stopped the selection procedure if none of the pathogens in the candidate list would increase the number of distinct combinations by at least two.
The simulations we conducted indicated further scrutiny was needed of pathogens exhibiting any of the following features in ANISA: a) high background noise (positive rate in healthy individuals larger than 10%); b) low resolution in detecting infection of the pathogen (odds ratio between cases and healthy individual s smaller than 1.0); and c) significant and substantial pairwise correlations of test results among healthy individuals (odds ratio larger than 2.0 or smaller than 0.5, with p-value smaller than 0.01), with possibility of excluding them from pLCM. Before exclusion, we examined the above features by key strata such as study site, age at enrollment, and age groups. We excluded pathogens only from the strata where the features were exhibited. We also examined possible correlation among pathogens. We used the information from this process to select or combine pathogen classes and create new combined tests for the new pathogen class.
While we were reluctant to apply informative prior distributions to either the pathogen proportion or the true positive rates for the ANISA analysis, we found that incorporating well-established knowledge was useful in improving the pLCMs performance. For example, the laboratory procedure (TAC) used in ANISA has very high laboratory sensitives for most of the pathogens (nearly 100%). Therefore, the true positive rates, approximately equal to the probability of the pathogen existing in the collected specimen multiplied by the laboratory sensitivity, conditional on cases being truly infected by the pathogen, should also be high. However, instead of assigning them some Beta distribution priors, we set lower limits to them: 40% for respiratory TAC and 20% for blood TAC tests. No lower limits were set for blood culture true positive rates due to lack of prior knowledge on blood culture test characteristics. The ANISA analysis is stratified by study site and disease severity status. The basic pLCM model can be easily extended for such stratification: we let the pathogen proportions and false positive rates change over the strata, but keep the true positive unchanged.
In pLCM, there is a reassignment of case and non-case status in each iteration of the Gibbs sampler. Therefore, for study designs with information from only a small sample of non-cases, cases that are not positive for a given pathogen in a given iteration can serve as non-cases and contribute to the estimation of the false positive rate for that pathogen. It is important to note that there may be a subset of cases who should never be considered as non-cases regardless of test results, e.g. because of very severe illness presentation or death.

![]()

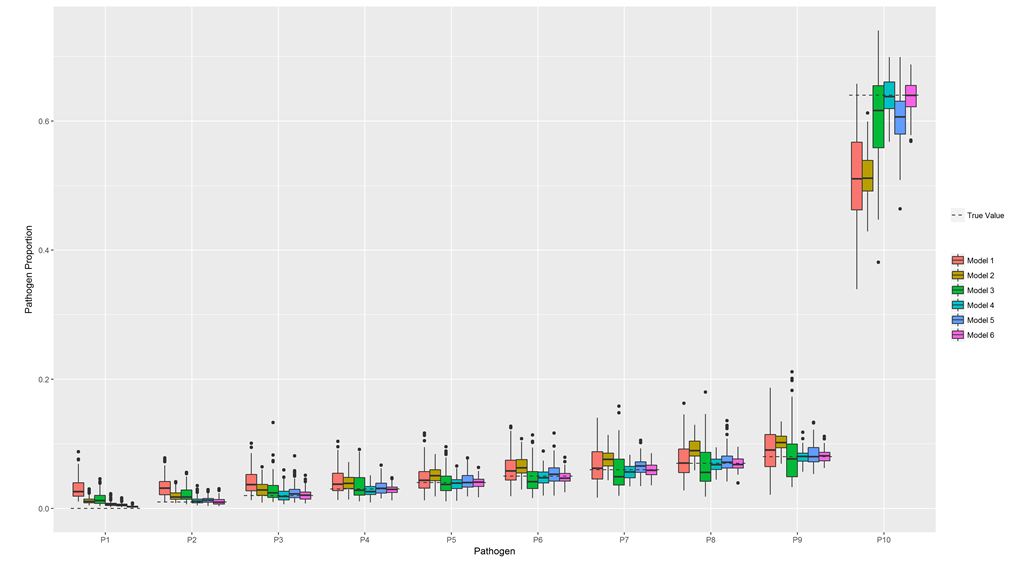 |
| Figure 1: Comparison of the accuracy of pathogen proportion estimates resulting from 6 different pLCM models using simulated data for ten pathogen classes Note: Three tests were simulated. Test 1: TPR=0.4, FPR=0.1; Test 2: TPR=0.85, FPR=0.1; and Test 3: TPR=0.2, FPR=0. Six models are fitted: Model 1: Test 1; Model 2: Test 2; Model 3: Test 1 and Test 3; Model 4: Test 2 and Test 3; Model 5: Test 1 and Test 2; Model 6: Test 1, Test 2, Test3. Based on 100 replicates of simulation runs. Standard boxplot structure is used displaying interquartile range, minimum and maximum values, and outliers. |
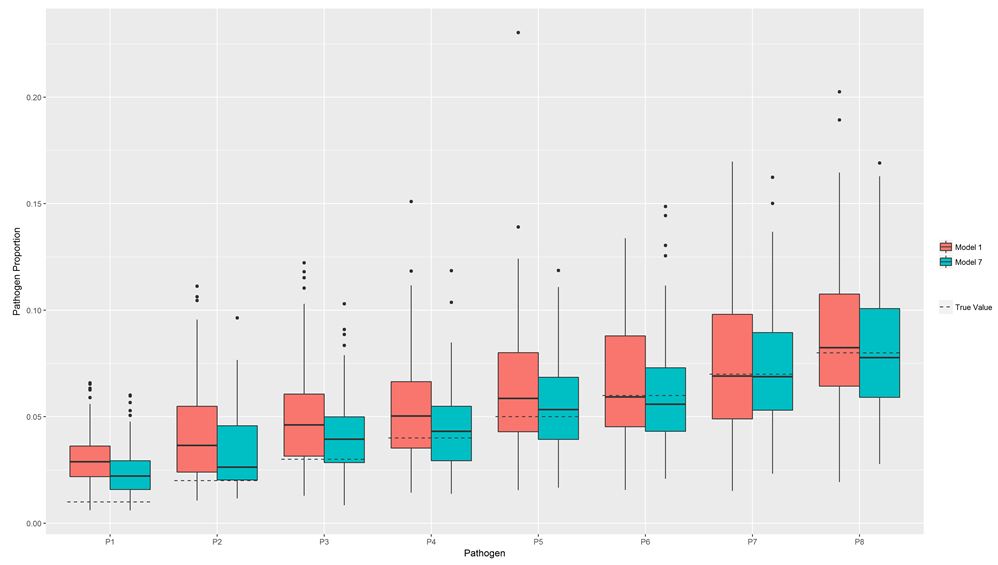 |
| Figure 2: Pathogen proportion estimate accuracy based on simulations of a pLCM model including 8 pathogen classes with a single test each (Model 1) and a model that includes an additional 8 pathogen classes with 3 tests each to the original 8 pathogens (Model 2) |
|
|
Pathogen |
|||||||||
True FPR |
True TPR |
P0 |
P1 |
P2 |
P3 |
P4 |
P5 |
P6 |
P7 |
P8 |
Other/None |
True pathogen proportion |
|||||||||||
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
64 |
||
|
|
Estimate |
|||||||||
0.3 |
0.4 |
8.43 |
8.37 |
9.24 |
8.86 |
8.66 |
8.97 |
8.87 |
9.14 |
9.18 |
20.28 |
0.6 |
7.73 |
7.93 |
8.68 |
8.78 |
8.99 |
9.48 |
9.77 |
9.83 |
10.45 |
18.37 |
|
0.85 |
6.43 |
6.63 |
7.04 |
8 |
8.37 |
9.61 |
10.27 |
10.39 |
11.95 |
21.31 |
|
0.95 |
5.39 |
5.73 |
6.83 |
7.78 |
7.93 |
9.07 |
9.54 |
10.84 |
12.07 |
24.83 |
|
0.1 |
0.4 |
4.92 |
5.65 |
5.59 |
6.21 |
6.98 |
7.04 |
8.01 |
9.04 |
8.81 |
37.74 |
0.6 |
3.2 |
3.71 |
3.91 |
4.98 |
6.27 |
6.25 |
6.84 |
8.39 |
9.12 |
47.34 |
|
0.85 |
2.12 |
2.99 |
3.57 |
4.37 |
5.76 |
6.44 |
7.67 |
8.98 |
9.76 |
48.33 |
|
0.95 |
2.18 |
2.98 |
3.48 |
4.79 |
5.55 |
6.64 |
7.91 |
9.28 |
10.6 |
46.59 |
|
0.01 |
0.4 |
1.06 |
1.58 |
1.99 |
3.09 |
3.39 |
4.4 |
5.25 |
6.11 |
7.46 |
65.67 |
0.6 |
0.78 |
1.37 |
2.1 |
3.29 |
4.22 |
5.18 |
6.29 |
7.31 |
8.03 |
61.43 |
|
0.85 |
0.58 |
1.34 |
2.35 |
3.67 |
4.69 |
5.92 |
7.11 |
8.44 |
9.74 |
56.16 |
|
0.95 |
0.52 |
1.48 |
2.5 |
3.94 |
5.05 |
6.21 |
7.54 |
8.97 |
10.27 |
53.52 |
|
|
|
Percent of replicates with absolute diff over true value > 0.5 |
|||||||||
0.3 |
0.4 |
|
100 |
100 |
100 |
89 |
68 |
41 |
32 |
12 |
|
0.6 |
|
100 |
100 |
100 |
92 |
69 |
57 |
37 |
27 |
|
|
0.85 |
|
100 |
98 |
95 |
71 |
65 |
63 |
52 |
47 |
|
|
0.95 |
|
100 |
95 |
84 |
67 |
60 |
50 |
48 |
48 |
|
|
0.1 |
0.4 |
|
100 |
93 |
71 |
56 |
44 |
36 |
30 |
24 |
|
0.6 |
|
96 |
67 |
58 |
45 |
34 |
21 |
21 |
20 |
|
|
0.85 |
|
91 |
59 |
37 |
44 |
30 |
29 |
35 |
27 |
|
|
0.95 |
|
85 |
53 |
45 |
33 |
36 |
34 |
33 |
33 |
|
|
0.01 |
0.4 |
|
51 |
19 |
25 |
16 |
8 |
7 |
6 |
5 |
|
0.6 |
|
38 |
20 |
14 |
15 |
8 |
3 |
1 |
1 |
|
|
0.85 |
|
38 |
25 |
19 |
16 |
7 |
7 |
4 |
4 |
|
|
0.95 |
|
43 |
33 |
26 |
16 |
15 |
11 |
9 |
7 |
|
|
|
Pathogen |
|||||||||||
|
P0 |
P1 |
P2 |
P3 |
P4 |
P5 |
P6 |
P7 |
P8 |
|||
Model |
True Pathogen Proportion |
|||||||||||
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
||||
|
Percent of replicates with absolute difference over true value>0.5 |
|||||||||||
1 |
100 |
97 |
63 |
41 |
28 |
29 |
30 |
22 |
27 |
|||
2 |
100 |
65 |
47 |
35 |
26 |
27 |
15 |
19 |
12 |
|||
3 |
100 |
67 |
40 |
37 |
23 |
23 |
23 |
29 |
25 |
|||
4 |
100 |
24 |
21 |
13 |
6 |
6 |
0 |
0 |
0 |
|||
5 |
100 |
31 |
25 |
11 |
11 |
11 |
4 |
9 |
4 |
|||
6 |
100 |
29 |
21 |
4 |
1 |
1 |
0 |
0 |
0 |
|||
|
Pathogen |
||||||||
P0 |
P1 |
P2 |
P3 |
P4 |
P5 |
P6 |
P7 |
P8 |
|
True Pathogen Proportion |
|||||||||
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Estimate (%) |
|||||||||
pLCM |
0.30 |
1.08 |
2.11 |
2.97 |
4.02 |
4.87 |
5.91 |
6.92 |
8.08 |
Regular latent class |
0.94 |
1.46 |
2.24 |
3.15 |
3.93 |
4.92 |
6.20 |
7.24 |
8.09 |
Root Mean Square Error (RMSE) |
|||||||||
pLCM |
0.34 |
0.56 |
0.88 |
0.68 |
0.88 |
0.99 |
0.91 |
1.04 |
0.99 |
Regular latent class |
1.21 |
0.99 |
1.31 |
1.58 |
1.55 |
2.00 |
1.99 |
2.43 |
2.08 |
RMSE over true value |
|||||||||
pLCM |
0.68 |
0.56 |
0.44 |
0.23 |
0.22 |
0.20 |
0.15 |
0.15 |
0.12 |
Regular latent class |
2.42 |
0.99 |
0.66 |
0.53 |
0.39 |
0.40 |
0.33 |
0.35 |
0.26 |
Percent of replicates with absolute difference |
|||||||||
pLCM |
100 |
29 |
21 |
4 |
2 |
1 |
0 |
0 |
0 |
Regular latent class |
100 |
33 |
33 |
33 |
19 |
18 |
14 |
11 |
5 |
|
Pathogen |
|||||||
P1 |
P2 |
P3 |
P4 |
P5 |
P6 |
P7 |
P8 |
|
True Pathogen Proportion |
||||||||
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
|
8 pathogens with a single test |
|||||||
Estimate (%) |
3.05 |
4.23 |
4.89 |
5.29 |
6.40 |
6.61 |
7.51 |
8.93 |
Root Mean Square Error (RMSE) |
2.42 |
3.19 |
3.06 |
2.75 |
3.51 |
3.02 |
3.44 |
3.40 |
RMSE over true value |
2.42 |
1.59 |
1.02 |
0.69 |
0.70 |
0.50 |
0.49 |
0.43 |
Percent of replicates with absolute difference |
100 |
64 |
54 |
42 |
34 |
32 |
37 |
20 |
Bias |
2.05 |
2.23 |
1.89 |
1.29 |
1.40 |
0.61 |
0.51 |
0.93 |
|
Addition of 8 pathogens with 3 tests each |
|||||||
Estimate (%) |
2.46 |
3.35 |
4.10 |
4.41 |
5.80 |
6.15 |
7.27 |
8.26 |
Root Mean Square Error (RMSE) |
1.84 |
2.23 |
2.12 |
1.96 |
2.46 |
2.58 |
2.83 |
2.90 |
RMSE over true value |
1.84 |
1.12 |
0.71 |
0.49 |
0.49 |
0.43 |
0.40 |
0.36 |
Percent of replicates with absolute difference |
100 |
44 |
40 |
26 |
25 |
18 |
17 |
12 |
Bias |
1.46 |
1.35 |
1.10 |
0.41 |
0.80 |
0.15 |
0.27 |
0.26 |




































